Differential Regulation of Cancer Progression by CDK4/6 Plays A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Screen of Cell-Cycle Regulators in Normal and Tumor Cells
bioRxiv preprint doi: https://doi.org/10.1101/060350; this version posted June 23, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion Maria Sokolova1, Mikko Turunen1, Oliver Mortusewicz3, Teemu Kivioja1, Patrick Herr3, Anna Vähärautio1, Mikael Björklund1, Minna Taipale2, Thomas Helleday3 and Jussi Taipale1,2,* 1Genome-Scale Biology Program, P.O. Box 63, FI-00014 University of Helsinki, Finland. 2Science for Life laboratory, Department of Biosciences and Nutrition, Karolinska Institutet, SE- 141 83 Stockholm, Sweden. 3Science for Life laboratory, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, S-171 21 Stockholm, Sweden To identify cell cycle regulators that enable cancer cells to replicate DNA and divide in an unrestricted manner, we performed a parallel genome-wide RNAi screen in normal and cancer cell lines. In addition to many shared regulators, we found that tumor and normal cells are differentially sensitive to loss of the histone genes transcriptional regulator CASP8AP2. In cancer cells, loss of CASP8AP2 leads to a failure to synthesize sufficient amount of histones in the S-phase of the cell cycle, resulting in slowing of individual replication forks. Despite this, DNA replication fails to arrest, and tumor cells progress in an elongated S-phase that lasts several days, finally resulting in death of most of the affected cells. -

HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap18559c
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP18559c Specification HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) - Product Information Application WB,E Primary Accession Q93079 Other Accession P57053, Q9PSW9, P0C1H5, P0C1H4, Q6PC60, Q8CGP0, Q8N257, Q9D2U9, Q5QNW6, Q64524, Q16778, Q64525, Q00715, Q5BJA5, P0C1H3, P62808, Q8CGP2, P23527, Q99877, Q32L48, P10854, Q99879, Q99880, Q8CGP1, O60814, Q2M2T1, HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody P06899, Q64478, (Center) (Cat. #AP18559c) western blot P10853, P58876, analysis in MCF-7 cell line lysates Q6ZWY9, P62807 (35ug/lane).This demonstrates the Reactivity Human HIST1H2BH/HIST1H2BK/HIST3H2BB antibody Predicted Xenopus, Mouse, detected the Bovine, Chicken, HIST1H2BH/HIST1H2BK/HIST3H2BB protein Zebrafish, Rat (arrow). Host Rabbit Clonality Polyclonal Isotype Rabbit Ig HIST1H2BH/HIST1H2BK/HIST3H2BB Calculated MW 13892 Antibody (Center) - Background Antigen Region 26-52 Histones are basic nuclear proteins that are HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody responsible (Center) - Additional Information for the nucleosome structure of the chromosomal fiber in Gene ID 8345 eukaryotes. Two molecules of each of the four core histones (H2A, Other Names H2B, H3, and H4) form an octamer, around Histone H2B type 1-H, Histone H2Bj, H2B/j, which approximately 146 bp HIST1H2BH, H2BFJ of DNA is wrapped in repeating units, called nucleosomes. The Target/Specificity linker histone, H1, interacts with linker DNA This HIST1H2BH/HIST1H2BK/HIST3H2BB between nucleosomes antibody is generated from rabbits and functions in the compaction of chromatin immunized with a KLH conjugated synthetic into higher order peptide between 26-52 amino acids from structures. This gene is intronless and encodes the Central region of human a member of the HIST1H2BH/HIST1H2BK/HIST3H2BB. -
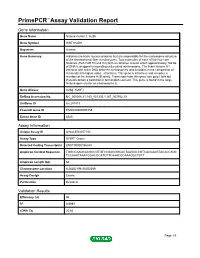
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 1, H2bh Gene Symbol HIST1H2BH Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A H2B H3 and H4) form an octamer around which approximately 146 bp of DNA is wrapped in repeating units called nucleosomes. The linker histone H1 interacts with linker DNA between nucleosomes and functions in the compaction of chromatin into higher order structures. This gene is intronless and encodes a member of the histone H2B family. Transcripts from this gene lack polyA tails but instead contain a palindromic termination element. This gene is found in the large histone gene cluster on chromosome 6. Gene Aliases H2B/j, H2BFJ RefSeq Accession No. NC_000006.11, NG_001335.1, NT_007592.15 UniGene ID Hs.247815 Ensembl Gene ID ENSG00000197459 Entrez Gene ID 8345 Assay Information Unique Assay ID qHsaCED0057192 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000356350 Amplicon Context Sequence TGGCCAAGCACGCCGTGTCCGAGGGCACTAAGGCCGTCACCAAGTACACCAGC TCCAAATAAATGGACGCATGTTCAAACCCAAAGGCTCTT Amplicon Length (bp) 62 Chromosome Location 6:26252198-26252289 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9997 cDNA Cq 20.62 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 81 gDNA Cq 25.12 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

The in Vitro and in Vivo Effects of Re-Expressing Methylated Von Hippel-Lindau Tumor Suppressor Gene in Clear Cell Renal Carcinoma with 5-Aza-2-Deoxycytidine
Vol. 10, 7011–7021, October 15, 2004 Clinical Cancer Research 7011 The In vitro and In vivo Effects of Re-Expressing Methylated von Hippel-Lindau Tumor Suppressor Gene in Clear Cell Renal Carcinoma with 5-Aza-2-deoxycytidine Wade G. Alleman,1,2 Ray L. Tabios,2 Well described phenotypic changes of VHL expression in- Gadisetti V. R. Chandramouli,3 cluding decreased invasiveness into Matrigel, and decreased Olga N. Aprelikova,3 Carlos Torres-Cabala,2 vascular endothelial growth factor and glucose transport- 4 5 er-1 expression were observed in the treated lines. VHL Arnulfo Mendoza, Craig Rodgers, methylated ccRCC xenografted tumors were significantly 2 2 Nikolai A. Sopko, W. Marston Linehan, and reduced in size in mice treated with 5-aza-dCyd. Mice bear- James R. Vasselli2 ing nonmethylated but VHL-mutated tumors showed no 1Howard Hughes Medical Institute, Chevy Chase, MD; 2Urology tumor shrinkage with 5-aza-dCyd treatment. Branch, 3Laboratory of Biosystems and Cancer, and 4Pediatric Conclusion: Hypo-methylating agents may be useful in Oncology Branch, Center for Cancer Research, National Cancer 5 the treatment of patients having ccRCC tumors consisting of Institute, Bethesda, Maryland; The Brady Urologic Institute, The cells with methylated VHL. Johns Hopkins Hospital, Baltimore, Maryland INTRODUCTION ABSTRACT An estimated 31,900 people are diagnosed annually with Purpose: Clear cell renal carcinoma (ccRCC) is cancer of the kidney in the United States, of which the majority strongly associated with loss of the von Hippel-Lindau (VHL) are clear cell type. In sporadic clear cell renal carcinoma tumor suppressor gene. The VHL gene is functionally lost (ccRCC), 50–85% of patients are found to have biallelic loss of through hypermethylation in up to 19% of sporadic ccRCC the von Hippel-Lindau (VHL) tumor suppressor gene (1–3), cases. -

Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands That Promote Axonal Growth
Research Article: New Research Development Peripheral Nerve Single-Cell Analysis Identifies Mesenchymal Ligands that Promote Axonal Growth Jeremy S. Toma,1 Konstantina Karamboulas,1,ª Matthew J. Carr,1,2,ª Adelaida Kolaj,1,3 Scott A. Yuzwa,1 Neemat Mahmud,1,3 Mekayla A. Storer,1 David R. Kaplan,1,2,4 and Freda D. Miller1,2,3,4 https://doi.org/10.1523/ENEURO.0066-20.2020 1Program in Neurosciences and Mental Health, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada, 2Institute of Medical Sciences University of Toronto, Toronto, Ontario M5G 1A8, Canada, 3Department of Physiology, University of Toronto, Toronto, Ontario M5G 1A8, Canada, and 4Department of Molecular Genetics, University of Toronto, Toronto, Ontario M5G 1A8, Canada Abstract Peripheral nerves provide a supportive growth environment for developing and regenerating axons and are es- sential for maintenance and repair of many non-neural tissues. This capacity has largely been ascribed to paracrine factors secreted by nerve-resident Schwann cells. Here, we used single-cell transcriptional profiling to identify ligands made by different injured rodent nerve cell types and have combined this with cell-surface mass spectrometry to computationally model potential paracrine interactions with peripheral neurons. These analyses show that peripheral nerves make many ligands predicted to act on peripheral and CNS neurons, in- cluding known and previously uncharacterized ligands. While Schwann cells are an important ligand source within injured nerves, more than half of the predicted ligands are made by nerve-resident mesenchymal cells, including the endoneurial cells most closely associated with peripheral axons. At least three of these mesen- chymal ligands, ANGPT1, CCL11, and VEGFC, promote growth when locally applied on sympathetic axons. -

Cell Cycle Arrest Through Indirect Transcriptional Repression by P53: I Have a DREAM
Cell Death and Differentiation (2018) 25, 114–132 Official journal of the Cell Death Differentiation Association OPEN www.nature.com/cdd Review Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM Kurt Engeland1 Activation of the p53 tumor suppressor can lead to cell cycle arrest. The key mechanism of p53-mediated arrest is transcriptional downregulation of many cell cycle genes. In recent years it has become evident that p53-dependent repression is controlled by the p53–p21–DREAM–E2F/CHR pathway (p53–DREAM pathway). DREAM is a transcriptional repressor that binds to E2F or CHR promoter sites. Gene regulation and deregulation by DREAM shares many mechanistic characteristics with the retinoblastoma pRB tumor suppressor that acts through E2F elements. However, because of its binding to E2F and CHR elements, DREAM regulates a larger set of target genes leading to regulatory functions distinct from pRB/E2F. The p53–DREAM pathway controls more than 250 mostly cell cycle-associated genes. The functional spectrum of these pathway targets spans from the G1 phase to the end of mitosis. Consequently, through downregulating the expression of gene products which are essential for progression through the cell cycle, the p53–DREAM pathway participates in the control of all checkpoints from DNA synthesis to cytokinesis including G1/S, G2/M and spindle assembly checkpoints. Therefore, defects in the p53–DREAM pathway contribute to a general loss of checkpoint control. Furthermore, deregulation of DREAM target genes promotes chromosomal instability and aneuploidy of cancer cells. Also, DREAM regulation is abrogated by the human papilloma virus HPV E7 protein linking the p53–DREAM pathway to carcinogenesis by HPV.Another feature of the pathway is that it downregulates many genes involved in DNA repair and telomere maintenance as well as Fanconi anemia. -

Identification of a Histone Family Gene Signature for Predicting The
www.nature.com/scientificreports OPEN Identifcation of a histone family gene signature for predicting the prognosis of cervical cancer Received: 21 August 2017 Accepted: 13 November 2017 patients Published: xx xx xxxx Xiaofang Li1, Run Tian2, Hugh Gao 3,4, Yongkang Yang1, Bryan R. G. Williams 3,4, Michael P. Gantier 3,4, Nigel A. J. McMillan5, Dakang Xu3,4,6, Yiqun Hu6 & Yan’e Gao1 Heterogeneity in terms of tumor characteristics, prognosis, and survival among cancer patients is an unsolved issue. Here, we systematically analyzed the aberrant expression patterns of cervical cancer using RNA-Seq data from The Cancer Genome Atlas (TCGA). We incorporated gene profling, molecular signatures, functional and pathway information with gene set enrichment and protein- protein interaction (PPI) network analysis, to identify sub-networks of genes. Those identifed genes relating to DNA replication and DNA repair-mediated signaling pathways associated with systemic lupus erythematosus (SLE). Next, we combined cross-validated prognostic scores to build an integrated prognostic model for survival prediction. The combined approach revealed that the DNA repair- mediated including the functional interaction module of 18 histone genes (Histone cluster 1 H2A, B and H4), were signifcantly correlated with the survival rate. Furthermore, fve of these histone genes were highly expressed in three cervical cancer cohorts from the Oncomine database. Comparison of high and low histone variant-expressing human cervical cancer cell lines revealed diferent responses to DNA damage, suggesting protective functions of histone genes against DNA damage. Collectively, we provide evidence that two SLE-associated gene sets (HIST1H2BD and HIST1H2BJ; and HIST1H2BD, HIST1H2BJ, HIST1H2BH, HIST1H2AM and HIST1H4K) can be used as prognostic factors for survival prediction among cervical cancer patients. -

PDF Datasheet
Product Datasheet HIST1H2BH Overexpression Lysate NBP2-07989 Unit Size: 0.1 mg Store at -80C. Avoid freeze-thaw cycles. Protocols, Publications, Related Products, Reviews, Research Tools and Images at: www.novusbio.com/NBP2-07989 Updated 3/17/2020 v.20.1 Earn rewards for product reviews and publications. Submit a publication at www.novusbio.com/publications Submit a review at www.novusbio.com/reviews/destination/NBP2-07989 Page 1 of 2 v.20.1 Updated 3/17/2020 NBP2-07989 HIST1H2BH Overexpression Lysate Product Information Unit Size 0.1 mg Concentration The exact concentration of the protein of interest cannot be determined for overexpression lysates. Please contact technical support for more information. Storage Store at -80C. Avoid freeze-thaw cycles. Buffer RIPA buffer Target Molecular Weight 13.7 kDa Product Description Description Transient overexpression lysate of histone cluster 1, H2bh (HIST1H2BH) The lysate was created in HEK293T cells, using Plasmid ID RC216657 and based on accession number NM_003524. The protein contains a C-MYC/DDK Tag. Gene ID 8345 Gene Symbol HIST1H2BH Species Human Notes HEK293T cells in 10-cm dishes were transiently transfected with a non-lipid polymer transfection reagent specially designed and manufactured for large volume DNA transfection. Transfected cells were cultured for 48hrs before collection. The cells were lysed in modified RIPA buffer (25mM Tris-HCl pH7.6, 150mM NaCl, 1% NP-40, 1mM EDTA, 1xProteinase inhibitor cocktail mix, 1mM PMSF and 1mM Na3VO4, and then centrifuged to clarify the lysate. Protein concentration was measured by BCA protein assay kit.This product is manufactured by and sold under license from OriGene Technologies and its use is limited solely for research purposes. -

Introduction to Gene Set Analysis Lecture (Pdf)
Extracting Biological Information from Gene Lists Simon Andrews, Laura Biggins, Boo Virk [email protected] [email protected] v2020-10 Analysis of processed Biological Sample for sample: Data acquisition – material analysis sequencing, microarray analysis, mass spectrometry Isolation of Sample DNA, RNA or processing proteins What does this mean??? Raw data Results Table file(s) Containing hits – genes, transcripts or proteins Public databases Data analysis: identification of genes, transcripts or proteins Biological themes are not always obvious from gene lists Relate the hits to existing knowledge Descriptions aren’t always informative Gene Description Gpr55 G protein-coupled receptor 55 [Source:MGI Symbol;Acc:MGI:2685064] Ncl nucleolin [Source:MGI Symbol;Acc:MGI:97286] Aspm asp (abnormal spindle)-like, microcephaly associated (Drosophila) [Source:MGI Symbol;Acc:MGI:1334448] Tnfsf4 tumor necrosis factor (ligand) superfamily, member 4 [Source:MGI Symbol;Acc:MGI:104511] Ephx1 epoxide hydrolase 1, microsomal [Source:MGI Symbol;Acc:MGI:95405] Setx senataxin [Source:MGI Symbol;Acc:MGI:2443480] Angptl2 angiopoietin-like 2 [Source:MGI Symbol;Acc:MGI:1347002] Ggta1 glycoprotein galactosyltransferase alpha 1, 3 [Source:MGI Symbol;Acc:MGI:95704] Dab2ip disabled homolog 2 (Drosophila) interacting protein [Source:MGI Symbol;Acc:MGI:1916851] Neb nebulin [Source:MGI Symbol;Acc:MGI:97292] Ermn ermin, ERM-like protein [Source:MGI Symbol;Acc:MGI:1925017] Ckap5 cytoskeleton associated protein 5 [Source:MGI Symbol;Acc:MGI:1923036] Prr5l proline rich 5 like [Source:MGI Symbol;Acc:MGI:1919696] Arhgap11a Rho GTPase activating protein 11A [Source:MGI Symbol;Acc:MGI:2444300] Bub1b budding uninhibited by benzimidazoles 1 homolog, beta (S. -

Early Vertebrate Whole Genome Duplications Were Predated by a Period of Intense Genome Rearrangement
Downloaded from genome.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Early vertebrate whole genome duplications were predated by a period of intense genome rearrangement Andrew L. Hufton1, Detlef Groth1,2, Martin Vingron1, Hans Lehrach1, Albert J. Poustka1, Georgia Panopoulou1* 1. Max Planck for Molecular Genetics, Ihnestr. 73, 12169 Berlin, Germany. 2. Potsdam University, Bioinformatics Group, c/o Max Planck Institute of Molecular Plant Physiology, Am Muehlenberg 1, D-14476 Potsdam-Golm, Germany * Corresponding author: Max-Planck Institut für Molekulare Genetik, Ihnestrasse 73, D- 14195 Berlin Germany. email: [email protected], Tel: +49-30-84131235, Fax: +49- 30-84131128 Running title: Early vertebrate genome duplications and rearrangements Keywords: synteny, amphioxus, genome duplications, rearrangement rate, genome instability Downloaded from genome.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Hufton et al. Abstract Researchers, supported by data from polyploid plants, have suggested that whole genome duplication (WGD) may induce genomic instability and rearrangement, an idea which could have important implications for vertebrate evolution. Benefiting from the newly released amphioxus genome sequence (Branchiostoma floridae), an invertebrate which researchers have hoped is representative of the ancestral chordate genome, we have used gene proximity conservation to estimate rates of genome rearrangement throughout vertebrates and some of their invertebrate ancestors. We find that, while amphioxus remains the best single source of invertebrate information about the early chordate genome, its genome structure is not particularly well conserved and it cannot be considered a fossilization of the vertebrate pre- duplication genome. In agreement with previous reports, we identify two WGD events in early vertebrates and another in teleost fish. -

PDF Download
Histone H2B (Tri Methyl Lys43) Polyclonal Antibody Catalog No : YM3290 Reactivity : Human,Mouse,Rat Applications : WB Gene Name : HIST1H2BC Protein Name : Histone H2B type 1-A/Histone H2B type 1-B/Histone H2B type 1-C/E/F/G/I Human Gene Id : 255626/3018/3017/8339/8343/8344/8346/8347 Human Swiss Prot Q96A08/P33778/P62807 No : Mouse Gene Id : 319177/319178/319179 Rat Gene Id : 24829 Rat Swiss Prot No : Q00729 Immunogen : Synthetic Peptide of Histone H2B (Tri Methyl Lys43) Specificity : The antibody detects endogenous Histone H2B (Tri Methyl Lys43) protein. Formulation : PBS, pH 7.4, containing 0.5%BSA, 0.02% sodium azide as Preservative and 50% Glycerol. Source : Rabbit Dilution : WB: 1:500-1000 Purification : The antibody was affinity-purified from rabbit antiserum by affinity- chromatography using specific immunogen. Storage Stability : -20°C/1 year Molecularweight : 14167/13950/13906 1 / 3 Observed Band : 14 Cell Pathway : Systemic lupus erythematosus, Background : histone cluster 1 H2B family member a(HIST1H2BA) Homo sapiens Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Nucleosomes consist of approximately 146 bp of DNA wrapped around a histone octamer composed of pairs of each of the four core histones (H2A, H2B, H3, and H4). The chromatin fiber is further compacted through the interaction of a linker histone, H1, with the DNA between the nucleosomes to form higher order chromatin structures. This gene is intronless and encodes a replication-dependent histone that is a testis/sperm-specific member of the histone H2B family. Transcripts from this gene contain a palindromic termination element. -

HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody Cat
HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody Cat. No.: 59-972 HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human Predicted species reactivity based on immunogen sequence: Xenopus, Mouse, Bovine, HOMOLOGY: Chicken, Zebrafish, Rat This HIST1H2BH/HIST1H2BK/HIST3H2BB antibody is generated from rabbits immunized IMMUNOGEN: with a KLH conjugated synthetic peptide between 26-52 amino acids from the Central region of human HIST1H2BH/HIST1H2BK/HIST3H2BB. TESTED APPLICATIONS: WB APPLICATIONS: For WB starting dilution is: 1:1000 PREDICTED MOLECULAR 14 kDa WEIGHT: Properties This antibody is purified through a protein A column, followed by peptide affinity PURIFICATION: purification. CLONALITY: Polyclonal September 26, 2021 1 https://www.prosci-inc.com/hist1h2bh-hist1h2bk-hist3h2bb-antibody-59-972.html ISOTYPE: Rabbit Ig CONJUGATE: Unconjugated PHYSICAL STATE: Liquid BUFFER: Supplied in PBS with 0.09% (W/V) sodium azide. CONCENTRATION: batch dependent Store at 4˚C for three months and -20˚C, stable for up to one year. As with all antibodies STORAGE CONDITIONS: care should be taken to avoid repeated freeze thaw cycles. Antibodies should not be exposed to prolonged high temperatures. Additional Info OFFICIAL SYMBOL: HIST1H2BH ALTERNATE NAMES: Histone H2B type 1-H, Histone H2Bj, H2B/j, HIST1H2BH, H2BFJ ACCESSION NO.: Q93079 PROTEIN GI NO.: 7387739 GENE ID: 8345 USER NOTE: Optimal dilutions for each application to be determined by the researcher. Background and References Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A, H2B, H3, and H4) form an octamer, around which approximately 146 bp of DNA is wrapped in repeating units, called nucleosomes.