Pdf (Accessed on 3 May 2018)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genome-Wide Screen of Cell-Cycle Regulators in Normal and Tumor Cells
bioRxiv preprint doi: https://doi.org/10.1101/060350; this version posted June 23, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion Maria Sokolova1, Mikko Turunen1, Oliver Mortusewicz3, Teemu Kivioja1, Patrick Herr3, Anna Vähärautio1, Mikael Björklund1, Minna Taipale2, Thomas Helleday3 and Jussi Taipale1,2,* 1Genome-Scale Biology Program, P.O. Box 63, FI-00014 University of Helsinki, Finland. 2Science for Life laboratory, Department of Biosciences and Nutrition, Karolinska Institutet, SE- 141 83 Stockholm, Sweden. 3Science for Life laboratory, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, S-171 21 Stockholm, Sweden To identify cell cycle regulators that enable cancer cells to replicate DNA and divide in an unrestricted manner, we performed a parallel genome-wide RNAi screen in normal and cancer cell lines. In addition to many shared regulators, we found that tumor and normal cells are differentially sensitive to loss of the histone genes transcriptional regulator CASP8AP2. In cancer cells, loss of CASP8AP2 leads to a failure to synthesize sufficient amount of histones in the S-phase of the cell cycle, resulting in slowing of individual replication forks. Despite this, DNA replication fails to arrest, and tumor cells progress in an elongated S-phase that lasts several days, finally resulting in death of most of the affected cells. -

A Strategic Research Alliance: Turner Syndrome and Sex Differences
A strategic research alliance: Turner syndrome and sex differences The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Roman, Adrianna K. San and David C. Page. “A strategic research alliance: Turner syndrome and sex differences.” American journal of medical genetics. Part C, Seminars in medical genetics 181 (2019): 59-67 © 2019 The Author(s) As Published 10.1002/AJMG.C.31677 Publisher Wiley Version Author's final manuscript Citable link https://hdl.handle.net/1721.1/125103 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ HHS Public Access Author manuscript Author ManuscriptAuthor Manuscript Author Am J Med Manuscript Author Genet C Semin Manuscript Author Med Genet. Author manuscript; available in PMC 2019 March 12. Published in final edited form as: Am J Med Genet C Semin Med Genet. 2019 March ; 181(1): 59–67. doi:10.1002/ajmg.c.31677. A strategic research alliance: Turner syndrome and sex differences Adrianna K. San Roman1 and David C. Page1,2,3 1Whitehead Institute, Cambridge, MA 02142, USA 2Howard Hughes Medical Institute, Whitehead Institute, Cambridge, MA 02142 3Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 Abstract Sex chromosome constitution varies in the human population, both between the sexes (46,XX females and 46,XY males), and within the sexes (for example, 45,X and 46,XX females, and 47,XXY and 46,XY males). Coincident with this genetic variation are numerous phenotypic differences between males and females, and individuals with sex chromosome aneuploidy. -

Sprouty Proteins, Masterminds of Receptor Tyrosine Kinase Signaling
CORE Metadata, citation and similar papers at core.ac.uk Provided by RERO DOC Digital Library Angiogenesis (2008) 11:53–62 DOI 10.1007/s10456-008-9089-1 ORIGINAL PAPER Sprouty proteins, masterminds of receptor tyrosine kinase signaling Miguel A. Cabrita Æ Gerhard Christofori Received: 14 December 2007 / Accepted: 7 January 2008 / Published online: 25 January 2008 Ó Springer Science+Business Media B.V. 2008 Abstract Angiogenesis relies on endothelial cells prop- Abbreviations erly processing signals from growth factors provided in Ang Angiopoietin both an autocrine and a paracrine manner. These mitogens c-Cbl Cellular homologue of Casitas B-lineage bind to their cognate receptor tyrosine kinases (RTKs) on lymphoma proto-oncogene product the cell surface, thereby activating a myriad of complex EGF Epidermal growth factor intracellular signaling pathways whose outputs include cell EGFR EGF receptor growth, migration, and morphogenesis. Understanding how eNOS Endothelial nitric oxide synthase these cascades are precisely controlled will provide insight ERK Extracellular signal-regulated kinase into physiological and pathological angiogenesis. The FGF Fibroblast growth factor Sprouty (Spry) family of proteins is a highly conserved FGFR FGF receptor group of negative feedback loop modulators of growth GDNF Glial-derived neurotrophic factor factor-mediated mitogen-activated protein kinase (MAPK) Grb2 Growth factor receptor-bound protein 2 activation originally described in Drosophila. There are HMVEC Human microvascular endothelial cell four mammalian orthologs (Spry1-4) whose modulation of Hrs Hepatocyte growth factor-regulated tyrosine RTK-induced signaling pathways is growth factor – and kinase substrate cell context – dependant. Endothelial cells are a group of HUVEC Human umbilical vein endothelial cell highly differentiated cell types necessary for defining the MAPK Mitogen-activated protein kinase mammalian vasculature. -

T Cell Depending on the Differentiation State of the Dual
Dual Effects of Sprouty1 on TCR Signaling Depending on the Differentiation State of the T Cell This information is current as Heonsik Choi, Sung-Yup Cho, Ronald H. Schwartz and of September 29, 2021. Kyungho Choi J Immunol 2006; 176:6034-6045; ; doi: 10.4049/jimmunol.176.10.6034 http://www.jimmunol.org/content/176/10/6034 Downloaded from References This article cites 63 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/176/10/6034.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 29, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2006 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Dual Effects of Sprouty1 on TCR Signaling Depending on the Differentiation State of the T Cell1 Heonsik Choi,* Sung-Yup Cho,† Ronald H. Schwartz,* and Kyungho Choi2* Sprouty (Spry) is known to be a negative feedback inhibitor of growth factor receptor signaling through inhibition of the Ras/ MAPK pathway. -

HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # Ap18559c
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) Affinity Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP18559c Specification HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody (Center) - Product Information Application WB,E Primary Accession Q93079 Other Accession P57053, Q9PSW9, P0C1H5, P0C1H4, Q6PC60, Q8CGP0, Q8N257, Q9D2U9, Q5QNW6, Q64524, Q16778, Q64525, Q00715, Q5BJA5, P0C1H3, P62808, Q8CGP2, P23527, Q99877, Q32L48, P10854, Q99879, Q99880, Q8CGP1, O60814, Q2M2T1, HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody P06899, Q64478, (Center) (Cat. #AP18559c) western blot P10853, P58876, analysis in MCF-7 cell line lysates Q6ZWY9, P62807 (35ug/lane).This demonstrates the Reactivity Human HIST1H2BH/HIST1H2BK/HIST3H2BB antibody Predicted Xenopus, Mouse, detected the Bovine, Chicken, HIST1H2BH/HIST1H2BK/HIST3H2BB protein Zebrafish, Rat (arrow). Host Rabbit Clonality Polyclonal Isotype Rabbit Ig HIST1H2BH/HIST1H2BK/HIST3H2BB Calculated MW 13892 Antibody (Center) - Background Antigen Region 26-52 Histones are basic nuclear proteins that are HIST1H2BH/HIST1H2BK/HIST3H2BB Antibody responsible (Center) - Additional Information for the nucleosome structure of the chromosomal fiber in Gene ID 8345 eukaryotes. Two molecules of each of the four core histones (H2A, Other Names H2B, H3, and H4) form an octamer, around Histone H2B type 1-H, Histone H2Bj, H2B/j, which approximately 146 bp HIST1H2BH, H2BFJ of DNA is wrapped in repeating units, called nucleosomes. The Target/Specificity linker histone, H1, interacts with linker DNA This HIST1H2BH/HIST1H2BK/HIST3H2BB between nucleosomes antibody is generated from rabbits and functions in the compaction of chromatin immunized with a KLH conjugated synthetic into higher order peptide between 26-52 amino acids from structures. This gene is intronless and encodes the Central region of human a member of the HIST1H2BH/HIST1H2BK/HIST3H2BB. -

Transcriptomic and Epigenomic Characterization of the Developing Bat Wing
ARTICLES OPEN Transcriptomic and epigenomic characterization of the developing bat wing Walter L Eckalbar1,2,9, Stephen A Schlebusch3,9, Mandy K Mason3, Zoe Gill3, Ash V Parker3, Betty M Booker1,2, Sierra Nishizaki1,2, Christiane Muswamba-Nday3, Elizabeth Terhune4,5, Kimberly A Nevonen4, Nadja Makki1,2, Tara Friedrich2,6, Julia E VanderMeer1,2, Katherine S Pollard2,6,7, Lucia Carbone4,8, Jeff D Wall2,7, Nicola Illing3 & Nadav Ahituv1,2 Bats are the only mammals capable of powered flight, but little is known about the genetic determinants that shape their wings. Here we generated a genome for Miniopterus natalensis and performed RNA-seq and ChIP-seq (H3K27ac and H3K27me3) analyses on its developing forelimb and hindlimb autopods at sequential embryonic stages to decipher the molecular events that underlie bat wing development. Over 7,000 genes and several long noncoding RNAs, including Tbx5-as1 and Hottip, were differentially expressed between forelimb and hindlimb, and across different stages. ChIP-seq analysis identified thousands of regions that are differentially modified in forelimb and hindlimb. Comparative genomics found 2,796 bat-accelerated regions within H3K27ac peaks, several of which cluster near limb-associated genes. Pathway analyses highlighted multiple ribosomal proteins and known limb patterning signaling pathways as differentially regulated and implicated increased forelimb mesenchymal condensation in differential growth. In combination, our work outlines multiple genetic components that likely contribute to bat wing formation, providing insights into this morphological innovation. The order Chiroptera, commonly known as bats, is the only group of To characterize the genetic differences that underlie divergence in mammals to have evolved the capability of flight. -
Primepcr™Assay Validation Report
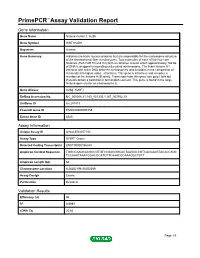
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 1, H2bh Gene Symbol HIST1H2BH Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A H2B H3 and H4) form an octamer around which approximately 146 bp of DNA is wrapped in repeating units called nucleosomes. The linker histone H1 interacts with linker DNA between nucleosomes and functions in the compaction of chromatin into higher order structures. This gene is intronless and encodes a member of the histone H2B family. Transcripts from this gene lack polyA tails but instead contain a palindromic termination element. This gene is found in the large histone gene cluster on chromosome 6. Gene Aliases H2B/j, H2BFJ RefSeq Accession No. NC_000006.11, NG_001335.1, NT_007592.15 UniGene ID Hs.247815 Ensembl Gene ID ENSG00000197459 Entrez Gene ID 8345 Assay Information Unique Assay ID qHsaCED0057192 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000356350 Amplicon Context Sequence TGGCCAAGCACGCCGTGTCCGAGGGCACTAAGGCCGTCACCAAGTACACCAGC TCCAAATAAATGGACGCATGTTCAAACCCAAAGGCTCTT Amplicon Length (bp) 62 Chromosome Location 6:26252198-26252289 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9997 cDNA Cq 20.62 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 81 gDNA Cq 25.12 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Microrna21 Contributes to Myocardial Disease by Stimulating MAP Kinase
Vol 456 | 18/25 December 2008 | doi:10.1038/nature07511 LETTERS MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts Thomas Thum1,2*, Carina Gross3*, Jan Fiedler1,2, Thomas Fischer3, Stephan Kissler3, Markus Bussen5, Paolo Galuppo1, Steffen Just6, Wolfgang Rottbauer6, Stefan Frantz1, Mirco Castoldi7,8,Ju¨rgen Soutschek9, Victor Koteliansky10, Andreas Rosenwald4, M. Albert Basson11, Jonathan D. Licht12, John T. R. Pena13, Sara H. Rouhanifard13, Martina U. Muckenthaler7,8, Thomas Tuschl13, Gail R. Martin5, Johann Bauersachs1 & Stefan Engelhardt3,14 MicroRNAs comprise a broad class of small non-coding RNAs that fibroblasts; expression was highest in fibroblasts from the failing control expression of complementary target messenger RNAs1,2. heart, but was low in cardiomyocytes (Fig. 2c and data not shown). Dysregulation of microRNAs by several mechanisms has been 3–5 6–10 a Early stage Intermediate stage Late stage described in various disease states including cardiac disease . 2 ) Whereas previous studies of cardiac disease have focused on 2 microRNAs that are primarily expressed in cardiomyocytes, the 1 role of microRNAs expressed in other cell types of the heart is 0 unclear. Here we show that microRNA-21 (miR-21, also known as Mirn21) regulates the ERK–MAP kinase signalling pathway in –1 control mice (log control failure model versus failure cardiac fibroblasts, which has impacts on global cardiac structure miRNA level in heart –2 and function. miR-21 levels are increased selectively in fibroblasts *** of the failing heart, augmenting ERK–MAP kinase activity through b Non-failing inhibition of sprouty homologue 1 (Spry1). This mechanism reg- 6 Failing ulates fibroblast survival and growth factor secretion, apparently Non-failing Failing *** controlling the extent of interstitial fibrosis and cardiac hyper- Early miR-21 4 trophy. -

NIH Public Access Author Manuscript Hepatology
NIH Public Access Author Manuscript Hepatology. Author manuscript; available in PMC 2012 December 11. Published in final edited form as: Hepatology. 2007 August ; 46(2): 548–557. doi:10.1002/hep.21682. Genome-level analysis of genetic regulation of liver gene expression networks $watermark-text $watermark-text $watermark-text Daniel Gatti1,¶, Akira Maki1,¶, Elissa J. Chesler2,¶, Roumyana Kirova2, Oksana Kosyk1, Lu Lu3, Kenneth F. Manly, Yanhua Qu3, Robert W. Williams3, Andy Perkins4, Michael A. Langston4, David W. Threadgill5,&, and Ivan Rusyn1,&,* Daniel Gatti: [email protected]; Akira Maki: [email protected]; Elissa J. Chesler: [email protected]; Roumyana Kirova: [email protected]; Oksana Kosyk: [email protected]; Lu Lu: [email protected]; Kenneth F. Manly: [email protected]; Yanhua Qu: [email protected]; Robert W. Williams: [email protected]; Andy Perkins: [email protected]; Michael A. Langston: [email protected]; David W. Threadgill: [email protected] 1Department of Environmental Sciences and Engineering, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 2Life Sciences Division, Oak Ridge National Laboratory, P.O. Box 2008, Oak Ridge, TN 37831 3Department of Anatomy and Neurobiology, University of Tennessee Health Science Center, Memphis, TN 38163 4Department of Computer Science, University of Tennessee, Knoxville, TN 37996 5Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 Abstract Liver is the primary site for metabolism of nutrients, drugs and chemical agents. While metabolic pathways are complex and tightly regulated, genetic variation among individuals, reflected in variation in gene expression levels, introduces complexity into research on liver disease. -

Thesis Resubmission Daphne Chen
Integrative Modelling of Glucocorticoid Induced Apoptosis with a Systems Biology Approach A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Sciences 2012 Daphne Wei-Chen Chen 1 Table of Contents List of Abbreviations ..................................................................................................................... 7 List of Tables ................................................................................................................................ 11 List of Figures .............................................................................................................................. 12 Abstract ........................................................................................................................................ 14 Short Abstract ............................................................................................................................. 15 Declaration ................................................................................................................................... 15 Copyright Statement ................................................................................................................... 15 Acknowledgements ...................................................................................................................... 16 1 CHAPTER 1: Introduction .......................................................................................... 17 1.1 Glucocorticoids (GCs) ................................................................................................. -

Meta-Analysis of Gene Expression in Individuals with Autism Spectrum Disorders
Meta-analysis of Gene Expression in Individuals with Autism Spectrum Disorders by Carolyn Lin Wei Ch’ng BSc., University of Michigan Ann Arbor, 2011 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Master of Science in THE FACULTY OF GRADUATE AND POSTDOCTORAL STUDIES (Bioinformatics) The University of British Columbia (Vancouver) August 2013 c Carolyn Lin Wei Ch’ng, 2013 Abstract Autism spectrum disorders (ASD) are clinically heterogeneous and biologically complex. State of the art genetics research has unveiled a large number of variants linked to ASD. But in general it remains unclear, what biological factors lead to changes in the brains of autistic individuals. We build on the premise that these heterogeneous genetic or genomic aberra- tions will converge towards a common impact downstream, which might be reflected in the transcriptomes of individuals with ASD. Similarly, a considerable number of transcriptome analyses have been performed in attempts to address this question, but their findings lack a clear consensus. As a result, each of these individual studies has not led to any significant advance in understanding the autistic phenotype as a whole. The goal of this research is to comprehensively re-evaluate these expression profiling studies by conducting a systematic meta-analysis. Here, we report a meta-analysis of over 1000 microarrays across twelve independent studies on expression changes in ASD compared to unaffected individuals, in blood and brain. We identified a number of genes that are consistently differentially expressed across studies of the brain, suggestive of effects on mitochondrial function. In blood, consistent changes were more difficult to identify, despite individual studies tending to exhibit larger effects than the brain studies.