Signalling Interactions Between Platelets and Lymphatic Endothelial
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Te2, Part Iii
TERMINOLOGIA EMBRYOLOGICA Second Edition International Embryological Terminology FIPAT The Federative International Programme for Anatomical Terminology A programme of the International Federation of Associations of Anatomists (IFAA) TE2, PART III Contents Caput V: Organogenesis Chapter 5: Organogenesis (continued) Systema respiratorium Respiratory system Systema urinarium Urinary system Systemata genitalia Genital systems Coeloma Coelom Glandulae endocrinae Endocrine glands Systema cardiovasculare Cardiovascular system Systema lymphoideum Lymphoid system Bibliographic Reference Citation: FIPAT. Terminologia Embryologica. 2nd ed. FIPAT.library.dal.ca. Federative International Programme for Anatomical Terminology, February 2017 Published pending approval by the General Assembly at the next Congress of IFAA (2019) Creative Commons License: The publication of Terminologia Embryologica is under a Creative Commons Attribution-NoDerivatives 4.0 International (CC BY-ND 4.0) license The individual terms in this terminology are within the public domain. Statements about terms being part of this international standard terminology should use the above bibliographic reference to cite this terminology. The unaltered PDF files of this terminology may be freely copied and distributed by users. IFAA member societies are authorized to publish translations of this terminology. Authors of other works that might be considered derivative should write to the Chair of FIPAT for permission to publish a derivative work. Caput V: ORGANOGENESIS Chapter 5: ORGANOGENESIS -

The Evolving Cardiac Lymphatic Vasculature in Development, Repair and Regeneration
REVIEWS The evolving cardiac lymphatic vasculature in development, repair and regeneration Konstantinos Klaourakis 1,2, Joaquim M. Vieira 1,2,3 ✉ and Paul R. Riley 1,2,3 ✉ Abstract | The lymphatic vasculature has an essential role in maintaining normal fluid balance in tissues and modulating the inflammatory response to injury or pathogens. Disruption of normal development or function of lymphatic vessels can have severe consequences. In the heart, reduced lymphatic function can lead to myocardial oedema and persistent inflammation. Macrophages, which are phagocytic cells of the innate immune system, contribute to cardiac development and to fibrotic repair and regeneration of cardiac tissue after myocardial infarction. In this Review, we discuss the cardiac lymphatic vasculature with a focus on developments over the past 5 years arising from the study of mammalian and zebrafish model organisms. In addition, we examine the interplay between the cardiac lymphatics and macrophages during fibrotic repair and regeneration after myocardial infarction. Finally, we discuss the therapeutic potential of targeting the cardiac lymphatic network to regulate immune cell content and alleviate inflammation in patients with ischaemic heart disease. The circulatory system of vertebrates is composed of two after MI. In this Review, we summarize the current complementary vasculatures, the blood and lymphatic knowledge on the development, structure and function vascular systems1. The blood vasculature is a closed sys- of the cardiac lymphatic vasculature, with an emphasis tem responsible for transporting gases, fluids, nutrients, on breakthroughs over the past 5 years in the study of metabolites and cells to the tissues2. This extravasation of cardiac lymphatic heterogeneity in mice and zebrafish. -

Patent Ductus Arteriosus About This Factsheet the Normal Heart
Understanding your child’s heart Patent ductus arteriosus About this factsheet The normal heart This factsheet is for parents of babies and children who The heart is a muscular pump which pumps blood through the have patent ductus arteriosus (PDA), which is also known as body and lungs. There are four chambers in the heart. The two persistent arterial duct. upper ones are called the right atrium and left atrium. These are separated by a wall called the atrial septum. The two lower It explains: chambers are called the right and left ventricles, and are separated • what patent ductus arteriosus is and how it is diagnosed by a wall called the ventricular septum. • how patent ductus arteriosus is treated • the benefits and risks of treatments. On each side of the heart, blood passes from the atrium, through a heart valve – the tricuspid valve on the right, and the mitral valve This factsheet does not replace the advice that doctors or on the left – into the ventricle. The ventricles are the main pumping nurses may give you, but it should help you to understand chambers of the heart. Each ventricle pumps blood out into an artery. what they tell you. The right ventricle pumps blood – blue in the illustration – into the pulmonary artery (the blood vessel that takes blood to the lungs). The left ventricle pumps blood – red in the illustration – into the aorta (the blood vessel that takes blood to the rest of the body). Blood flows from the right side of the heart, through the pulmonary valve into the pulmonary artery, and then to the lungs where it picks up oxygen. -
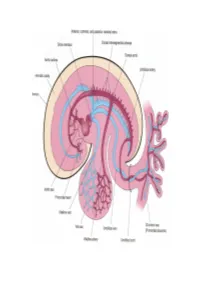
Development of HEART 4-VEINS
Development of brachiocephalic veins 1. Right brachiocephalic vein is formed by cranial part of right anterior cardinal vein and 2. Left brachiocephalic is formed by cranial part of left anterior cardinal vein and the interant.cardinal anastomosis. Development of superior vena cava 1. The part up to the opening of vena azygos develops from caudal part of right ant.cardinal vein and 2. The part below the opening (intrapericardial part) is formed by the right common cardinal vein. Development of azygos and hemiazygos veins A. 1. Vena azygos develops from right azygos line vein and 2. The arch of vena azygos is formed by the cranial end of right postcardinal vein. B. Hemiazygos veins are formed by the left azygos line vein. Development of Inferior vena cava Inferior vena cava is formed, from below upwards by: 1. Begins by the union of the two common iliac veins (postcardinal veins), 2. Right supracardinal, 3. Right supra-subcardinal anastomosis, 4. Right subcardinal, 5. New formation (hepatic segment) and 6. Hepatocardiac channel (terminal part of right vitelline vein). Congenital anomalies • Double inferior vena cava • Absence • Left SVC • Double SVC DEVELOPMENT OF PORTAL VEIN 1. The portal vein is formed behind the neck of pancreas by the union of superior mesentric and splenic vein to the left vitelline vein. 2. The part of the portal vein which is behind the Ist part of duodenum is formed by middle dorsal transverse anastomosis. 3. Part of portal vein which is in the free margin of lesser omentum is formed by cranial or distal part of right vitelline vein. -

Lymphangiogenesis and Angiogenesis During Human Fetal
Roost et al. Vascular Cell 2014, 6:22 http://www.vascularcell.com/content/6/1/22 VASCULAR CELL RESEARCH Open Access Lymphangiogenesis and angiogenesis during human fetal pancreas development Matthias S Roost1, Liesbeth van Iperen1, Ana de Melo Bernardo1, Christine L Mummery1, Françoise Carlotti2, Eelco JP de Koning2,3 and Susana M Chuva de Sousa Lopes1,4* Abstract Background: The complex endocrine and exocrine functionality of the human pancreas depends on an efficient fluid transport through the blood and the lymphatic vascular systems. The lymphatic vasculature has key roles in the physiology of the pancreas and in regulating the immune response, both important for developing successful transplantation and cell-replacement therapies to treat diabetes. However, little is known about how the lymphatic and blood systems develop in humans. Here, we investigated the establishment of these two vascular systems in human pancreas organogenesis in order to understand neovascularization in the context of emerging regenerative therapies. Methods: We examined angiogenesis and lymphangiogenesis during human pancreas development between 9 and 22 weeks of gestation (W9-W22) by immunohistochemistry. Results: As early as W9, the peri-pancreatic mesenchyme was populated by CD31-expressing blood vessels as well as LYVE1- and PDPN-expressing lymphatic vessels. The appearance of smooth muscle cell-coated blood vessels in the intra-pancreatic mesenchyme occurred only several weeks later and from W14.5 onwards the islets of Langerhans also became heavily irrigated by blood vessels. In contrast to blood vessels, LYVE1- and PDPN-expressing lymphatic vessels were restricted to the peri-pancreatic mesenchyme until later in development (W14.5-W17), and some of these invading lymphatic vessels contained smooth muscle cells at W17. -

The Rediscovery of the Lymphatic System: Old and New Insights Into the Development and Biological Function of the Lymphatic Vasculature
Downloaded from genesdev.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW The rediscovery of the lymphatic system: old and new insights into the development and biological function of the lymphatic vasculature Guillermo Oliver1,3 and Michael Detmar2,3 1Department of Genetics, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, USA; 2Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts 02114, USA The lymphatic system is composed of a vascular net- control lymphatic development and function. These work of thin-walled capillaries that drain protein-rich findings include the identification of specific genetic de- lymph from the extracellular spaces within most organs. fects in certain hereditary diseases that are associated A continuous single-cell layer of overlapping endothelial with lymphatic hypoplasia and dysfunction (i.e., lymph- cells lines the lymphatic capillaries, which lack a con- edemas; Milroy 1892; Meige 1898), and evidence that tinuous basement membrane and are, therefore, highly malignant tumors can directly activate lymphangiogen- permeable. Lymph returns to venous circulation via the esis and lymphatic metastasis (Karpanen et al. 2001; larger lymphatic collecting vessels, which contain a Mandriota et al. 2001; Skobe et al. 2001a; Stacker et al. muscular and adventitial layer, and the thoracic duct. 2001). The lymphatic system also includes lymphoid organs such as the lymph nodes, tonsils, Peyer’s patches, spleen, -

Fetal Circulation
The Fetal Circulation Dr. S. Mathieu, Specialist Registrar in Anaesthesia Dr. D. J. Dalgleish, Consultant Anaesthetist Royal Bournemouth and Christchurch Hospitals Trust, UK Questions 1. In the fetal circulation: a) There are two umbilical arteries and one umbilical vein? b) Over 90% of blood passes the liver via the ductus venosus c) The foramen ovale divides the left and right ventricle d) The umbilical artery carries oxygenated blood from the placenta to the fetus e) The foramen ovale allows oxygenated blood to bypass the pulmonary circulation 2. In the fetal circulation: a) The oxygen dissociation curve of fetal haemoglobin is shifted to the left compared with adult haemoglobin ensuring oxygen delivery to the fetus despite low oxygen partial pressures b) It is the presence of the ductus arteriosus and large pulmonary vascular resistance which ensures most of the right ventricular output passes into the aorta c) The patency of the ductus arteriosus is maintained by high oxygen tensions d) The patency of the ductus arteriosus is maintained by the vasodilating effects of prostaglandin G2 e) 2,3-DPG levels are higher in fetal haemoglobin compared with adult haemaglobin 3. Changes at birth include: a) a fall in pulmonary vascular resistance b) a rise in systemic vascular resistance with clamping of the cord c) an increase in hypoxic pulmonary vasoconstriction d) a rise in left atrial pressure e) closure of the ductus arteriosus within 24 hours 4. The following congenital heart lesions are cyanotic: a) Ventricular septal defect b) Atrial septal defect c) Patent ductus arteriosus d) Tetralogy of Fallot e) Transposition of the great arteries MCQ answers at end Key points • The fetal circulation supplies the fetal tissues with oxygen and nutrients from the placenta. -

The Pattern and Mechanisms of Response to Oxygen by the Ductus Arteriosus and Umbilical Artery
Pediat. Res. 6: 693-700 (1972) Acetylcholine neonate atropine prematurity bradykinin sympathetic nervous system ductus arteriosus The Pattern and Mechanisms of Response to Oxygen by the Ductus Arteriosus and Umbilical Artery INGRID OBERHANSLI-WEISS, MICHAEL A. HEYMANN1391, ABRAHAM M. RUDOLPH, AND KENNETH L. MELMON Cardiovascular Research Institute and Departments of Pediatrics, Physiology and Pharmacology, University of California San Francisco, San Francisco, California, USA Extract Response of the ductus arteriosus and umbilical artery to changes in oxygen tension, to acetylcholine, and to sympathetic and parasympathetic blocking agents was studied in vitro in isolated rings obtained from 22 fetal lambs of 98- to 147-day gestation. After stabilization of tension at a baseline level (0.3-0.7 g) in a PO2 environment of 35-45 mm Hg, both increase of the PO2 to 550 mm Hg and decrease of the PO2 to 8 mm Hg of the bathing solution produced constriction. The mean maximal tension developed by the ductus arteriosus.was 3.91 g at high PO2 and 3.87 g at low POr The increase in maximal tension developed with advancing gestation was also similar at both high and low POj. At P02 levels of 8-550 mm Hg, acetylcholine produced a further increase in tension, whereas bradykinin only produced an increase in tension at high PO2- Alpha and beta sympathetic blockade had no effect on the constrictor response to oxygen. Atropine relaxed the ductus arteriosus and umbilical artery at both high and low Po2 levels; the degree of relaxation was related to drug concentration. Acetylcholin- esterase also relaxed the ductus arteriosus constricted by oxygen. -

Echocardiographic Follow-Up of Patent Foramen Ovale and the Factors Affecting Spontaneous Closure
Acta Cardiol Sin 2016;32:731-737 Brief Report doi: 10.6515/ACS20160205A Echocardiographic Follow-Up of Patent Foramen Ovale and the Factors Affecting Spontaneous Closure Ali Yildirim,1 Alperen Aydin,2 Tevfik Demir,1 Fatma Aydin,2 Birsen Ucar1 and Zubeyir Kilic1 Background: The aim of the present study was to evaluate the echocardiographic follow-up of patent foramen ovale, which is considered a potential etiological factor in various diseases, and to determine the factors affecting spontaneous closure. Methods: Between January 2000 and June 2012, records of 918 patients with patent foramen ovale were retrospectively reviewed. Patency of less than 3 mm around the fossa ovalis is called patent foramen ovale. Patients with cyanotic congenital heart diseases, severe heart valve disorders and severe hemodynamic left to right shunts were excluded from the study. The patients were divided into three groups based on age; 1 day-1 monthingroup1,1month-12monthsingroup2,andmorethan12monthsingroup3. Results: Of the 918 patients, 564 (61.4%) had spontaneous closure, 328 (35.8%) had patent foramen ovale continued, 15 (1.6%) patients had patent foramen ovale enlarged to 3-5 mm, 6 patients were enlarged to 5-8 mm, and in one patient patent foramen ovale reached to more than 8 mm size. Defect was spontaneously closed in 65.9% of the patients in group 1, 66.7% of the patients in group 2, and 52.3% of the patients in group 3. There was a negative correlation between the age of diagnosis and spontaneous closure (p < 0.05). Gender, prematurity and coexisting malformations such as patent ductus arteriosus and atrial septal aneurysm did not have any effect on spontaneous closure of patent foramen ovale (p > 0.05). -

Has a Role in Cardiovascular and Placental Development and Is a Binding Partner of the Α4 Integrin
Abl-interactor-1 (Abi1) has a role in cardiovascular and placental development and is a binding partner of the α4 integrin Colleen Ringa,1, Mark H. Ginsbergb, Jacob Halingb, and Ann Marie Pendergasta,1 aDepartment of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC 27710; and bDepartment of Medicine, University of California at San Diego, La Jolla, CA 92093 Edited* by Stephen P. Goff, Columbia University College of Physicians and Surgeons, New York, NY, and approved November 30, 2010 (received for review August 19, 2010) Dynamic signals linking the actin cytoskeleton and cell adhesion properties among integrin subunits in that α4 predominantly receptors are essential for morphogenesis during development accumulates at the leading edge of migrating cells, rather than at and normal tissue homeostasis. Abi1 is a central regulator of actin focal adhesions. Moreover, α4 expression is associated with polymerization through interactions with multiple protein com- protrusive activity and enhanced cell migration; however, the plexes. However, the in vivo role of Abi1 remains to be defined. pathways that link α4 integrin to the actin-regulatory machinery The α4 integrin adhesion receptor is associated with enhanced at the leading edge have remained elusive. Here we identify Abi1 protrusive activity and regulation of directional cell migration. as a target of α4 integrin that positively regulates membrane Among integrin subunits, α4 exhibits unique properties in that it protrusion by promoting actin polymerization at sites of integrin predominantly accumulates at the leading edge of migrating cells; engagement. however, the pathways that link the actin-regulatory machinery to The Abi family proteins, Abi1 and Abi2, were originally id- α4 at the leading edge have remained elusive. -

Lymphatic Tissue Engineering and Regeneration Laura Alderfer1, Alicia Wei1 and Donny Hanjaya-Putra1,2,3,4,5,6*
Alderfer et al. Journal of Biological Engineering (2018) 12:32 https://doi.org/10.1186/s13036-018-0122-7 REVIEW Open Access Lymphatic Tissue Engineering and Regeneration Laura Alderfer1, Alicia Wei1 and Donny Hanjaya-Putra1,2,3,4,5,6* Abstract The lymphatic system is a major circulatory system within the body, responsible for the transport of interstitial fluid, waste products, immune cells, and proteins. Compared to other physiological systems, the molecular mechanisms and underlying disease pathology largely remain to be understood which has hindered advancements in therapeutic options for lymphatic disorders. Dysfunction of the lymphatic system is associated with a wide range of disease phenotypes and has also been speculated as a route to rescue healthy phenotypes in areas including cardiovascular disease, metabolic syndrome, and neurological conditions. This review will discuss lymphatic system functions and structure, cell sources for regenerating lymphatic vessels, current approaches for engineering lymphatic vessels, and specific therapeutic areas that would benefit from advances in lymphatic tissue engineering and regeneration. Keywords: Lymphangiogenesis, Tissue Engineering, Disease Modeling, Wound Healing, Lymphedema, Stem Cells, Biomaterials, Interstitial Fluid, Regeneration I. Introduction to the Lymphatic System and its role Interstitial fluid (IF) is a plasma filtrate that is generated Function by transcapillary filtration and is governed by Starling The lymphatic system is nearly ubiquitous in the human forces, the net difference between hydrostatic and body, present in all tissues except the epidermis, cartil- osmotic pressures, at the microcirculatory level [9]. In age, eye lens, cornea, retina, and bone marrow [1, 2]. order to maintain fluid homeostasis, lymph formation in The main functions of the lymphatic system include the initial lymphatic vessels must be balanced by the net fluid homeostasis and interstitial fluid drainage, immune flux of plasma being filtered out [4]. -

6 Development of the Great Vessels and Conduction Tissue
Development of the Great Vessels and Conduc6on Tissue Development of the heart fields • h:p://php.med.unsw.edu.au/embryology/ index.php?6tle=Advanced_-_Heart_Fields ! 2 Septa6on of the Bulbus Cordis Bulbus Cordis AV Canal Ventricle Looking at a sagital sec6on of the heart early in development the bulbus cordis is con6nuous with the ventricle which is con6nuous with the atria. As the AV canal shiOs to the right the bulbus move to the right as well. Septa6on of the Bulbus Cordis A P A P The next three slides make the point via cross sec6ons that the aorta and pulmonary arteries rotate around each other. This means the septum between them changes posi6on from superior to inferior as well. Septa6on of the Bulbus Cordis P A A P Septa6on of the Bulbus Cordis P A P A Migra6on of neural crest cells Neural crest cells migrate from the 3ed, 4th and 6th pharyngeal arches to form some of the popula6on of cells forming the aor6copulmonary septum. Septa6on of the Bulbus Cordis Truncal (Conal) Swellings Bulbus Cordis The cardiac jelly in the region of the truncus and conus adds the neural crest cells and expands as truncal swellings. Septa6on of the Bulbus Cordis Aorticopulmonary septum These swellings grow toward each other to meet and form the septum between the aorta and pulmonary artery. Aorta Pulmonary Artery Septa6on of the Bulbus Cordis Anterior 1 2 3 1 2 3 The aor6copulmonary septum then rotates as it moves inferiorly. However, the exact mechanism for that rota6on remains unclear. Septa6on of the Bulbus Cordis Aorta Pulmonary Artery Conal Ridges IV Foramen Membranous Muscular IV Endocarial Septum Interventricular Cushion Septum However, the aor6copulmonary septum must form properly for the IV septum to be completed.