The Crystal Structure of Warditei
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Svanbergite and Other Alunite Group Minerals in Advanced Argillic Altered Rocks from Chervena Mogila Ore Deposit, Central Sredno
СПИСАНИЕ НА БЪЛГАРСКОТО ГЕОЛОГИЧЕСКО ДРУЖЕСТВО, год. 79, кн. 3, 2018, с. 21–22 REVIEW OF THE BULGARIAN GEOLOGICAL SOCIETY, vol. 79, part 3, 2018, p. 21–22 National Conference with international participation “GEOSCIENCES 2018” Svanbergite and other alunite group minerals in advanced argillic altered rocks from Chervena Mogila ore deposit, Central Srednogorie Сванбергит и други минерали от алунитовата група в интензивно аргилизираните скали от находище Червена Могила, Централно Средногорие Atanas Hikov, Nadezhda Velinova Атанас Хиков, Надежда Велинова Geological Institute, Bulgarian Academy of Sciences, 1113 Sofia; E-mails: [email protected]; [email protected] Keywords: svanbergite, alunite, APS, advanced argillic alteration, geochemistry, Central Srednogorie. Introduction (secondary quartzites) and silicic (monoquartz) types have limited development. The following minerals Alunite and aluminium phosphate-sulphate (APS) participate in the mineral assemblages of AAA rocks: minerals (Stoffregen, Alpers, 1987) have been object kaolinite, dickite, pyrophyllite, sericite, alunite, topaz of increasing interest in the last 40 years. They are part and tourmaline (Popov et al., 1994). of the alunite supergroup (Jambor, 1999) with general This study confirms the limited development of formula DG3(TO4)2(OH,H2O)6, where D are large cat- AAA rocks. They are more widespread in the south- ions (K, Na, Ag, Ca, Sr, Ba, Pb, Bi, La, Ce, Nd) with east direction in the region of Varnishka Chukara, coordination number larger or equal to 9. G site is oc- where they are likely to form a lithocap. Alunite, cupied by cations in octahedral coordination (Al, Fe, dickite-diaspore-pyrophyllite, monoquartz and mixed Cu, Zn), and T site – by S, P, As in tetrahedral coor- types are distinguished while in the latter case there is dination. -
A Raman and Infrared Spectroscopic Analysis of the Phosphate Mineral
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 126 (2014) 164–169 Contents lists available at ScienceDirect Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy journal homepage: www.elsevier.com/locate/saa A Raman and infrared spectroscopic analysis of the phosphate mineral wardite NaAl3(PO4)2(OH)4Á2(H2O) from Brazil ⇑ Ray L. Frost a, , Ricardo Scholz b, Andrés López a, Cristiano Lana b, Yunfei Xi a a School of Chemistry, Physics and Mechanical Engineering, Science and Engineering Faculty, Queensland University of Technology, GPO Box 2434, Brisbane, Queensland 4001, Australia b Geology Department, School of Mines, Federal University of Ouro Preto, Campus Morro do Cruzeiro, Ouro Preto, MG 35400-00, Brazil highlights graphical abstract A wardite mineral sample from Lavra Da Ilha, Minas Gerais, Brazil was analysed. Using SEM with EDX and vibrational spectroscopy. The calculated formula is (Na0.97Ca0.03)R1.00Al3 (PO4)2(OH)4Á2(H2O). Observation of multiple bands supports the concept of non- equivalent phosphate units in the structure. article info abstract Article history: A wardite mineral sample from Lavra Da Ilha, Minas Gerais, Brazil has been examined by vibrational spec- Received 17 November 2013 troscopy. The mineral is unusual in that it belongs to a unique symmetry class, namely the tetragonal-tra- Received in revised form 7 January 2014 pezohedral group. The structure of wardite contains layers of corner-linked –OH bridged MO6 octahedra Accepted 2 February 2014 stacked along the tetragonal C-axis in a four-layer sequence and linked by PO groups. Consequentially Available online 15 February 2014 4 not all phosphate units are identical. -

Mineralogical Study of the Advanced Argillic Alteration Zone at the Konos Hill Mo–Cu–Re–Au Porphyry Prospect, NE Greece †
Article Mineralogical Study of the Advanced Argillic Alteration Zone at the Konos Hill Mo–Cu–Re–Au Porphyry Prospect, NE Greece † Constantinos Mavrogonatos 1,*, Panagiotis Voudouris 1, Paul G. Spry 2, Vasilios Melfos 3, Stephan Klemme 4, Jasper Berndt 4, Tim Baker 5, Robert Moritz 6, Thomas Bissig 7, Thomas Monecke 8 and Federica Zaccarini 9 1 Faculty of Geology & Geoenvironment, National and Kapodistrian University of Athens, 15784 Athens, Greece; [email protected] 2 Department of Geological and Atmospheric Sciences, Iowa State University, Ames, IA 50011, USA; [email protected] 3 Faculty of Geology, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece; [email protected] 4 Institut für Mineralogie, Westfälische Wilhelms-Universität Münster, 48149 Münster, Germany; [email protected] (S.K.); [email protected] (J.B.) 5 Eldorado Gold Corporation, 1188 Bentall 5 Burrard St., Vancouver, BC V6C 2B5, Canada; [email protected] 6 Department of Mineralogy, University of Geneva, CH-1205 Geneva, Switzerland; [email protected] 7 Goldcorp Inc., Park Place, Suite 3400-666, Burrard St., Vancouver, BC V6C 2X8, Canada; [email protected] 8 Center for Mineral Resources Science, Department of Geology and Geological Engineering, Colorado School of Mines, 1516 Illinois Street, Golden, CO 80401, USA; [email protected] 9 Department of Applied Geosciences and Geophysics, University of Leoben, Leoben 8700, Austria; [email protected] * Correspondence: [email protected]; Tel.: +30-698-860-8161 † The paper is an extended version of our paper published in 1st International Electronic Conference on Mineral Science, 16–21 July 2018. Received: 8 October 2018; Accepted: 22 October 2018; Published: 24 October 2018 Abstract: The Konos Hill prospect in NE Greece represents a telescoped Mo–Cu–Re–Au porphyry occurrence overprinted by deep-level high-sulfidation mineralization. -

A Vibrational Spectroscopic Study of the Phosphate Mineral Vantasselite
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 147 (2015) 185–192 Contents lists available at ScienceDirect Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy journal homepage: www.elsevier.com/locate/saa A vibrational spectroscopic study of the phosphate mineral vantasselite Al4(PO4)3(OH)3Á9H2O ⇑ Ray L. Frost a, , Ricardo Scholz b, Fernanda Maria Belotti c, Andrés López a, Frederick L. Theiss a a School of Chemistry, Physics and Mechanical Engineering, Science and Engineering Faculty, Queensland University of Technology, GPO Box 2434, Brisbane, Queensland 4001, Australia b Geology Department, School of Mines, Federal University of Ouro Preto, Campus Morro do Cruzeiro, Ouro Preto, MG 35,400-00, Brazil c Federal University of Itajubá, Campus Itabira, Itabira, MG 35,903-087, Brazil highlights graphical abstract We have studied the phosphate mineral vantasselite Al2(PO4)(OH)3Á3H2O. Using a combination of SEM with EDX and Raman and infrared spectroscopy. Chemical analysis shows a mineral containing Al, Fe and P. A comparison is made with other aluminum containing phosphate minerals. Vibrational spectroscopy offers a mechanism for the study of the molecular structure of vantasselite. article info abstract Article history: We have studied the phosphate mineral vantasselite Al4(PO4)3(OH)3Á9H2O using a combination of SEM Received 28 October 2014 with EDX and Raman and infrared spectroscopy. Qualitative chemical analysis shows Al, Fe and P. Received in revised form 15 January 2015 À1 3À Raman bands at 1013 and 1027 cm are assigned to the PO4 m1 symmetric stretching mode. The obser- Accepted 23 March 2015 vation of two bands suggests the non-equivalence of the phosphate units in the vantasselite structure. -

New Mineral Names*,†
American Mineralogist, Volume 106, pages 1360–1364, 2021 New Mineral Names*,† Dmitriy I. Belakovskiy1, and Yulia Uvarova2 1Fersman Mineralogical Museum, Russian Academy of Sciences, Leninskiy Prospekt 18 korp. 2, Moscow 119071, Russia 2CSIRO Mineral Resources, ARRC, 26 Dick Perry Avenue, Kensington, Western Australia 6151, Australia In this issue This New Mineral Names has entries for 11 new species, including 7 minerals of jahnsite group: jahnsite- (NaMnMg), jahnsite-(NaMnMn), jahnsite-(CaMnZn), jahnsite-(MnMnFe), jahnsite-(MnMnMg), jahnsite- (MnMnZn), and whiteite-(MnMnMg); lasnierite, manganflurlite (with a new data for flurlite), tewite, and wumuite. Lasnierite* the LA-ICP-MS analysis, but their concentrations were below detec- B. Rondeau, B. Devouard, D. Jacob, P. Roussel, N. Stephant, C. Boulet, tion limits. The empirical formula is (Ca0.59Sr0.37)Ʃ0.96(Mg1.42Fe0.54)Ʃ1.96 V. Mollé, M. Corre, E. Fritsch, C. Ferraris, and G.C. Parodi (2019) Al0.87(P2.99Si0.01)Ʃ3.00(O11.41F0.59)Ʃ12 based on 12 (O+F) pfu. The strongest lines of the calculated powder X-ray diffraction pattern are [dcalc Å (I%calc; Lasnierite, (Ca,Sr)(Mg,Fe)2Al(PO4)3, a new phosphate accompany- ing lazulite from Mt. Ibity, Madagascar: an example of structural hkl)]: 4.421 (83; 040), 3.802 (63, 131), 3.706 (100; 022), 3.305 (99; 141), characterization from dynamic refinement of precession electron 2.890 (90; 211), 2.781 (69; 221), 2.772 (67; 061), 2.601 (97; 023). It diffraction data on submicrometer sample. European Journal of was not possible to perform powder nor single-crystal X-ray diffraction Mineralogy, 31(2), 379–388. -

Fluorowardite, Naal3(PO4)2(OH)2F2·2H2O, the Fluorine Analog of Wardite from the Silver Coin Mine, Valmy, Nevada
American Mineralogist, Volume 99, pages 804–810, 2014 Fluorowardite, NaAl3(PO4)2(OH)2F22 Anthony R. KAmpf1,*, pAul m. AdAms2, RobeRt m. housley3 And GeoRGe R. RossmAn3 1Mineral Sciences Department, Natural History Museum of Los Angeles County, 900 Exposition Boulevard, Los Angeles, California 90007, U.S.A. 2126 South Helberta Avenue, #2, Redondo Beach, California 90277, U.S.A. 3Division of Geological and Planetary Sciences, California Institute of Technology, Pasadena, California 91125, U.S.A. AbstRAct Fluorowardite (IMA2012-016), NaAl3(PO4)2(OH)2F2·2H2O, the F analog of wardite, is a new mineral from the Silver Coin mine, Valmy, Iron Point district, Humboldt County, Nevada, U.S.A., where it oc- curs as a low-temperature secondary mineral in complex phosphate assemblages rich in Al, Na, and F. Fluorowardite forms colorless to white or cream-colored, tetragonal-pyramidal crystals up to 0.1 mm in diameter. The streak is white. Crystals are transparent to translucent, with vitreous to pearly luster. The Mohs hardness is about 5, the tenacity is brittle, the fracture is irregular, and crystals exhibit one perfect cleavage on {001}. The calculated density is 2.760 g/cm3 analyses (average of 8) provided: Na2O 6.27, CaO 1.74, MgO 0.42, Al2O3 35.21, Fe2O3 0.72, P2O5 32.49, As2O52O 13.35 (structure), total 94.74 wt%. The presence of H2O and OH and the absence of CO3 3+ anions) is: (Na0.87Ca0.13Mg0.04)(Al2.96Fe0.04)(P1.96As0.03)O8.12(OH)2.35F1.53·2H2O. Fluorowardite 3 is tetragonal, P41212, acV , and Z lines in the X-ray powder diffraction pattern are [dobsI)(hkl)]: 4.766(100)(004,103); 3.099(75) (211,203); 3.008(62)(115,212); 2.834(28)(204,213); 2.597(56)(205); 1.7628(32)(400,401); 1.6592(29) R1FoF) 62O) octahedra, PO4 tetrahedra, and NaO6(H2O)26 octahedra link by corner-sharing to form a square array. -

Roscherite-Group Minerals from Brazil
■ ■ Roscherite-Group Minerals yÜÉÅ UÜté|Ä Daniel Atencio* and José M.V. Coutinho Instituto de Geociências, Universidade de São Paulo, Rua do Lago, 562, 05508-080 – São Paulo, SP, Brazil. *e-mail: [email protected] Luiz A.D. Menezes Filho Rua Esmeralda, 534 – Prado, 30410-080 - Belo Horizonte, MG, Brazil. INTRODUCTION The three currently recognized members of the roscherite group are roscherite (Mn2+ analog), zanazziite (Mg analog), and greifensteinite (Fe2+ analog). These three species are monoclinic but triclinic variations have also been described (Fanfani et al. 1977, Leavens et al. 1990). Previously reported Brazilian occurrences of roscherite-group minerals include the Sapucaia mine, Lavra do Ênio, Alto Serra Branca, the Córrego Frio pegmatite, the Lavra da Ilha pegmatite, and the Pirineus mine. We report here the following three additional occurrences: the Pomarolli farm, Lavra do Telírio, and São Geraldo do Baixio. We also note the existence of a fourth member of the group, an as-yet undescribed monoclinic Fe3+-dominant species with higher refractive indices. The formulas are as follows, including a possible formula for the new species: Roscherite Ca2Mn5Be4(PO4)6(OH)4 • 6H2O Zanazziite Ca2Mg5Be4(PO4)6(OH)4 • 6H2O 2+ Greifensteinite Ca2Fe 5Be4(PO4)6(OH)4 • 6H2O 3+ 3+ Fe -dominant Ca2Fe 3.33Be4(PO4)6(OH)4 • 6H2O ■ 1 ■ Axis, Volume 1, Number 6 (2005) www.MineralogicalRecord.com ■ ■ THE OCCURRENCES Alto Serra Branca, Pedra Lavrada, Paraíba Unanalyzed “roscherite” was reported by Farias and Silva (1986) from the Alto Serra Branca granite pegmatite, 11 km southwest of Pedra Lavrada, Paraíba state, associated with several other phosphates including triphylite, lithiophilite, amblygonite, tavorite, zwieselite, rockbridgeite, huréaulite, phosphosiderite, variscite, cyrilovite and mitridatite. -
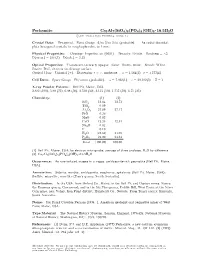
Perhamite Ca3al7(Sio4)3(PO4)4(OH)3 ² 16:5H2O C 2001 Mineral Data Publishing, Version 1.2 ° Crystal Data: Hexagonal
Perhamite Ca3Al7(SiO4)3(PO4)4(OH)3 ² 16:5H2O c 2001 Mineral Data Publishing, version 1.2 ° Crystal Data: Hexagonal. Point Group: 6=m 2=m 2=m (probable). As radial discoidal, platy hexagonal crystals, in rough spherules, to 1 mm. Physical Properties: Cleavage: Imperfect on 0001 . Tenacity: Brittle. Hardness = 5 f g » D(meas.) = 2.64(1) D(calc.) = 2.53 Optical Properties: Translucent to nearly opaque. Color: Brown, white. Streak: White. Luster: Dull, vitreous on cleavage surface. Optical Class: Uniaxial (+). Dispersion: r > v; moderate. ! = 1.564(2) ² = 1.577(4) Cell Data: Space Group: P 6=mmm (probable). a = 7.022(1) c = 20.182(5) Z = 1 X-ray Powder Pattern: Bell Pit, Maine, USA. 2.882 (100), 5.80 (71), 6.08 (50), 3.510 (50), 3.115 (50), 1.757 (50), 6.71 (35) Chemistry: (1) (2) SiO2 13.64 13.72 TiO2 0.09 Al2O3 27.09 27.17 FeO 0.26 MgO 0.02 CaO 12.26 12.81 Na2O 0.02 F 0.10 H2O [24.62] 24.69 P2O5 21.90 21.61 Total [100.00] 100.00 (1) Bell Pit, Maine, USA; by electron microprobe, average of three analyses, H2O by di®erence. (2) Ca3Al7(SiO4)3(PO4)4(OH)3 ² 16:5H2O Occurrence: As rare isolated masses in a vuggy, amblygonite-rich pegmatite (Bell Pit, Maine, USA). Association: Siderite, wardite, amblygonite, eosphorite, sphalerite (Bell Pit, Maine, USA); °uellite, minyulite, wavellite (Tom's quarry, South Australia). Distribution: In the USA, from Oxford Co., Maine, in the Bell Pit and Dunton mines, Newry; the Emmons quarry, Greenwood; and in the Ski Pike quarry, Cobble Hill, West Paris; at the Silver Coin mine, near Valmy, Iron Point district, Humboldt Co., Nevada. -

Fluorowardite Naal3(PO4)2(OH)2F2·2H2O
Fluorowardite NaAl3(PO4)2(OH)2F2·2H2O Crystal Data: Tetragonal. Point Group: 422. As tetragonal bipyramidal crystals, to 0.1 mm, displaying {001} (prominent and lustrous), {011} and/or {012} (prominent, irregular, and striated parallel to [100]), and {100} (common, irregular, and striated parallel to [100]). Physical Properties: Cleavage: Perfect on {001}. Fracture: Irregular. Tenacity: Brittle. Hardness = 5 D(meas.) = n.d. D(calc.) = 2.706 Optical Properties: Transparent to translucent. Color: Colorless to white or cream-colored. Streak: White. Luster: Vitreous to pearly. Optical Class: Uniaxial (+). ω = 1.576(2) ε = 1.584(2) Cell Data: Space Group: P41212. a = 7.077(2) c = 19.227(3) Z = 4 X-ray Powder Pattern: Silver Coin mine, Valmy, Humboldt County, Nevada, USA. 4.766 (100), 3.099 (75), 3.008 (62), 2.597 (56), 1.5228 (49), 1.7628 (32), 1.6592 (29) Chemistry: (1) (2) Na2O 6.27 7.71 CaO 1.74 MgO 0.42 Al2O3 35.21 38.05 Fe2O3 0.72 P2O5 32.49 35.32 As2O5 0.64 F 6.76 9.45 -O = F2 2.85 3.98 H2O [13.35] 13.45 total 94.74 100.00 (1) Silver Coin mine, Valmy, Humboldt County, Nevada, USA; average of 8 electron microprobe analyses supplemented by FTIR and Raman spectroscopy, H2O calculated from the structure; 3+ corresponds to (Na0.87Ca0.13Mg0.04)Σ=1.04(Al2.96Fe 0.04)Σ=3.00(P1.96As0.03)Σ=1.99O8.12(OH)2.35F1.53·2H2O. (2) NaAl3(PO4)2(OH)2F2·2H2O. Occurrence: A secondary phase, in a F-rich secondary phosphate assemblage. -

Aluminium Phosphates in Muscovite-Kyanite Metaquartzites from Passo Di Vizze (Alto Adige, NE Italy)
Eur. J. Mineral. 1996, 8, 853-869 Aluminium phosphates in muscovite-kyanite metaquartzites from Passo di Vizze (Alto Adige, NE Italy) GIULIO MORTEANI* and DIETRICH ACKERMAND** *Lehrstuhl für Angewandte Mineralogie, Technische Universität München, Lichtenbergstraße 4, D-85747 Garching, Germanyl **Mineralogisch-Petrographisches Institut, Universität Kiel, Olshausenstraße 40, D-24118 Kiel, Germany Abstract: A metaquartzite horizon bearing aluminium phosphates occurs over several kilometres in the Penninic Lower "Schieferhülle" units of the Tauem Window at the Passo di Vizze (Alto Adige, NE Italy). It contains the following types of mineral association: A) pale-blue lazulite, colourless crandallite-goyazite, apatite, tourmaline, B) dark-blue lazulite, colourless svanbergite-goyazite, bearthite, apatite, celestite, and C) staurolite, chlorite and tourmaline. Additional minerals in all cases are quartz, muscovite, kyanite and accessory Ti-bearing hematite. As sociation-types A and B are the common ones, type C is found only as a thin layer on the surface of coarse quartz pebbles embedded in types A or B. Staurolite is never found in contact with the phosphates. 2+ 2+ Electron microprobe analyses show that lazulite, (Mg,Fe )Ah(OH)2(P04h, has XMg [= Mg/(Mg+Fe )] ranging between 0.95 and 0.75 in association-type A, and between 0.65 and 0.55 in type B. (Sr,Ca)-bearing AI phosphates are a quatemary solid-solution series between crandallite-goyazite and woodhouseite-svanber gite (Ca,Sr)AbH1-x[(OH)6(P04h-x{(P04)(S04}x], and a binary one between bearthite and goedkenite, (Ca,SrhAI(OH)(P04h. The quatemary phase of type A has an XSr [= Sr/(Sr+Ca)] in the range of 0.35 to 0.95 and an Xp [=(P-I)/(P-l) +S] in the range of 0.45 to 0.90, indicating a main solid solution series between crandallite and goyazite; in the association-type B this phase has an XSr in the range of 0.95 to 0.99 and an Xp in the range of 0.29 to 0.90, showing a solid-solution series between svanbergite and goyazite. -

A Specific Gravity Index for Minerats
A SPECIFICGRAVITY INDEX FOR MINERATS c. A. MURSKyI ern R. M. THOMPSON, Un'fuersityof Bri.ti,sh Col,umb,in,Voncouver, Canad,a This work was undertaken in order to provide a practical, and as far as possible,a complete list of specific gravities of minerals. An accurate speciflc cravity determination can usually be made quickly and this information when combined with other physical properties commonly leads to rapid mineral identification. Early complete but now outdated specific gravity lists are those of Miers given in his mineralogy textbook (1902),and Spencer(M,i,n. Mag.,2!, pp. 382-865,I}ZZ). A more recent list by Hurlbut (Dana's Manuatr of M,i,neral,ogy,LgE2) is incomplete and others are limited to rock forming minerals,Trdger (Tabel,l,enntr-optischen Best'i,mmungd,er geste,i,nsb.ildend,en M,ineral,e, 1952) and Morey (Encycto- ped,iaof Cherni,cal,Technol,ogy, Vol. 12, 19b4). In his mineral identification tables, smith (rd,entifi,cati,onand. qual,itatioe cherai,cal,anal,ys'i,s of mineral,s,second edition, New york, 19bB) groups minerals on the basis of specificgravity but in each of the twelve groups the minerals are listed in order of decreasinghardness. The present work should not be regarded as an index of all known minerals as the specificgravities of many minerals are unknown or known only approximately and are omitted from the current list. The list, in order of increasing specific gravity, includes all minerals without regard to other physical properties or to chemical composition. The designation I or II after the name indicates that the mineral falls in the classesof minerals describedin Dana Systemof M'ineralogyEdition 7, volume I (Native elements, sulphides, oxides, etc.) or II (Halides, carbonates, etc.) (L944 and 1951). -

A Study of the Phosphate Mineral Kapundaite Naca(Fe3+)4(PO4)4
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 122 (2014) 400–404 Contents lists available at ScienceDirect Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy journal homepage: www.elsevier.com/locate/saa 3+ A study of the phosphate mineral kapundaite NaCa(Fe )4(PO4)4 (OH)3Á5(H2O) using SEM/EDX and vibrational spectroscopic methods ⇑ Ray L. Frost a, , Andrés López a, Yunfei Xi a, Ricardo Scholz b a School of Chemistry, Physics and Mechanical Engineering, Science and Engineering Faculty, Queensland University of Technology, GPO Box 2434, Brisbane, Queensland 4001, Australia b Geology Department, School of Mines, Federal University of Ouro Preto, Campus Morro do Cruzeiro, Ouro Preto, MG 35 400-00, Brazil highlights graphical abstract The kapundaite sample originated from the Sapucaia mine a lithium- bearing pegmatite. Kapundaite is a hydrated hydroxyl sodium calcium ferric phosphate 3+ NaCa(Fe )4(PO4)4(OH)3Á5(H2O). The molecular structure of kapundaite was assessed by vibrational spectroscopy. Multiple phosphate bands are observed. article info abstract Article history: Vibrational spectroscopy enables subtle details of the molecular structure of kapundaite to be deter- Received 17 September 2013 mined. Single crystals of a pure phase from a Brazilian pegmatite were used. Kapundaite is the Fe3+ mem- Received in revised form 5 November 2013 ber of the wardite group. The infrared and Raman spectroscopy were applied to compare the structure of Accepted 5 November 2013 kapundaite with wardite. The Raman spectrum of kapundaite in the 800–1400 cmÀ1 spectral range shows Available online 21 November 2013 À1 3À two intense bands at 1089 and 1114 cm assigned to the m1 PO4 symmetric stretching vibrations.