Retina II Jeanne L. Rosenthal MD MPOD FACS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Why Is This Important? Phone Triage) and Physical Exam
Goals Diagnostic Dilemmas Learn which features of the history and 1. What is the cause of this patient’s red eye? physical examination are most useful 2. Does this patient have a pathologic headache? Use risk scores and clinical decision tools 3. Does this patient have endocarditis? Distinguish benign from potentially serious 4. What is the cause of this patient’s back pain? conditions Identify clinical pearls and pitfalls What is the Cause of this Patient’s Mr. Ira Tatedi Red Eye? 32 year old man with one day h/o mild redness of OD, pain, and photophobia. Physical exam shows circumcorneal injection, and visual acuity is 20/80. Key Elements of History (including Why is this Important? Phone Triage) and Physical Exam Most cases of red eye are caused by viral History and Triage Physical Examination conjunctivitis, which does not generally Is vision affected? Visual acuity require any treatment Is there a foreign Pupil size/reactivity Some cases are caused by bacterial or allergic body sensation? Discharge conjunctivitis, for which specific treatment is Is there Pattern of redness indicated photophobia? Foreign body A minority of cases are caused by other Was there trauma? Hypopyon/hyphema conditions, which require urgent or emergent Are patient a contact referral to an ophthalmologist lens wearer? It is essential to be able to distinguish them Is there discharge from one another throughout the day? Anatomy/Differential Diagnosis Conjunctivitis Viral Conjunctivitis – Erythema with co-existing URI – Watery, serous discharge -
Differentiate Red Eye Disorders
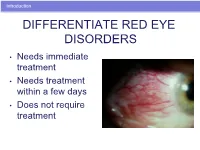
Introduction DIFFERENTIATE RED EYE DISORDERS • Needs immediate treatment • Needs treatment within a few days • Does not require treatment Introduction SUBJECTIVE EYE COMPLAINTS • Decreased vision • Pain • Redness Characterize the complaint through history and exam. Introduction TYPES OF RED EYE DISORDERS • Mechanical trauma • Chemical trauma • Inflammation/infection Introduction ETIOLOGIES OF RED EYE 1. Chemical injury 2. Angle-closure glaucoma 3. Ocular foreign body 4. Corneal abrasion 5. Uveitis 6. Conjunctivitis 7. Ocular surface disease 8. Subconjunctival hemorrhage Evaluation RED EYE: POSSIBLE CAUSES • Trauma • Chemicals • Infection • Allergy • Systemic conditions Evaluation RED EYE: CAUSE AND EFFECT Symptom Cause Itching Allergy Burning Lid disorders, dry eye Foreign body sensation Foreign body, corneal abrasion Localized lid tenderness Hordeolum, chalazion Evaluation RED EYE: CAUSE AND EFFECT (Continued) Symptom Cause Deep, intense pain Corneal abrasions, scleritis, iritis, acute glaucoma, sinusitis, etc. Photophobia Corneal abrasions, iritis, acute glaucoma Halo vision Corneal edema (acute glaucoma, uveitis) Evaluation Equipment needed to evaluate red eye Evaluation Refer red eye with vision loss to ophthalmologist for evaluation Evaluation RED EYE DISORDERS: AN ANATOMIC APPROACH • Face • Adnexa – Orbital area – Lids – Ocular movements • Globe – Conjunctiva, sclera – Anterior chamber (using slit lamp if possible) – Intraocular pressure Disorders of the Ocular Adnexa Disorders of the Ocular Adnexa Hordeolum Disorders of the Ocular -

Pattern of Vitreo-Retinal Diseases at the National Referral Hospital in Bhutan: a Retrospective, Hospital-Based Study Bhim B
Rai et al. BMC Ophthalmology (2020) 20:51 https://doi.org/10.1186/s12886-020-01335-x RESEARCH ARTICLE Open Access Pattern of vitreo-retinal diseases at the national referral hospital in Bhutan: a retrospective, hospital-based study Bhim B. Rai1,2* , Michael G. Morley3, Paul S. Bernstein4 and Ted Maddess1 Abstract Background: Knowing the pattern and presentation of the diseases is critical for management strategies. To inform eye-care policy we quantified the pattern of vitreo-retinal (VR) diseases presenting at the national referral hospital in Bhutan. Methods: We reviewed all new patients over three years from the retinal clinic of the Jigme Dorji Wangchuck National Referral Hospital. Demographic data, presenting complaints and duration, treatment history, associated systemic diseases, diagnostic procedures performed, and final diagnoses were quantified. Comparisons of the expected and observed frequency of gender used Chi-squared tests. We applied a sampling with replacement based bootstrap analysis (10,000 cycles) to estimate the population means and the standard errors of the means and standard error of the 10th, 25th, 50th, 75th and 90th percentiles of the ages of the males and females within 20-year cohorts. We then applied t-tests employing the estimated means and standard errors. The 2913 subjects insured that the bootstrap estimates were statistically conservative. Results: The 2913 new cases were aged 47.2 ± 21.8 years. 1544 (53.0%) were males. Housewives (953, 32.7%) and farmers (648, 22.2%) were the commonest occupations. Poor vision (41.9%), screening for diabetic and hypertensive retinopathy (13.1%), referral (9.7%), sudden vision loss (9.3%), and trauma (8.0%) were the commonest presenting symptoms. -

Frequency of Common Factors Leading to Leukocoria in Pediatric Age Group at Tertiary Care Eye Hospital: an Observational Study
Biostatistics and Biometrics Open Access Journal ISSN: 2573-2633 Research Article Biostat Biometrics Open Acc J Faisal’s Issue - January 2018 Copyright © All rights are reserved by Muhammad Faisal Fahim DOI: 10.19080/BBOAJ.2018.04.555636 Frequency of Common Factors Leading to Leukocoria in Pediatric Age Group at Tertiary Care Eye Hospital: An Observational Study Rizwana Dargahi1, Abdul Haleem1, Darikta Dargahi S1 and Muhammad Faisal Fahim2* 1Department of Ophthalmology, Shaheed Mohtarma Benazir Bhutto Medical University, Pakistan 2Department of Research & Development, Al-Ibrahim Eye Hospital, Pakistan Submission: November 27, 2017; Published: January 19, 2018 *Corresponding author: Muhammad Faisal Fahim, M.Sc. (Statistics), Statistician, Department of Research & Development, Al-Ibrahim Eye Hospital, Isra postgraduate Institute of Ophthalmology, Karachi, Pakistan, Tel: ; Email: Abstract Objective: To know the frequency of common factors leading to leukocoria in pediatric age group at a tertiary care eye hospital. Materials & methods: This was across sectional observational study carried out at Pediatric department, Al-Ibrahim eye hospital Karachi from February to July 2016. All patients younger than 10 years who presented with leukocoria were included. The excluding criteria were previous history of trauma and ocular surgery. Systemic as well as ocular examination and relevant investigations was done to know cause of was used to analyzed data. leukocoria. B. Scan was done in all the cases. If needed X-ray chest, CT scan & MRI orbit was done for confirming diagnosis. SPSS version 20.0 Results: In our country congenital cataract is the most common cause of leukocoria which comprises about 77.3% (133/172) which is treatable. 2nd common cause was persistent fetal vasculature comprises 8.1% (14/172). -

Strabismus, Amblyopia & Leukocoria
Strabismus, Amblyopia & Leukocoria [ Color index: Important | Notes: F1, F2 | Extra ] EDITING FILE Objectives: ➢ Not given. Done by: Jwaher Alharbi, Farrah Mendoza. Revised by: Rawan Aldhuwayhi Resources: Slides + Notes + 434 team. NOTE: F1& F2 doctors are different, the doctor who gave F2 said she is in the exam committee so focus on her notes Amblyopia ● Definition Decrease in visual acuity of one eye without the presence of an organic cause that explains that decrease in visual acuity. He never complaints of anything and his family never noticed any abnormalities ● Incidence The most common cause of visual loss under 20 years of life (2-4% of the general population) ● How? Cortical ignorance of one eye. This will end up having a lazy eye ● binocular vision It is achieved by the use of the two eyes together so that separate and slightly dissimilar images arising in each eye are appreciated as a single image by the process of fusion. It’s importance 1. Stereopsis 2. Larger field If there is no coordination between the two eyes the person will have double vision and confusion so as a compensatory mechanism for double vision the brain will cause suppression. The visual pathway is a plastic system that continues to develop during childhood until around 6-9 years of age. During this time, the wiring between the retina and visual cortex is still developing. Any visual problem during this critical period, such as a refractive error or strabismus can mess up this developmental wiring, resulting in permanent visual loss that can't be fixed by any corrective means when they are older Why fusion may fail ? 1. -

CAUSES, COMPLICATIONS &TREATMENT of A“RED EYE”
CAUSES, COMPLICATIONS & TREATMENT of a “RED EYE” 8 Most cases of “red eye” seen in general practice are likely to be conjunctivitis or a superficial corneal injury, however, red eye can also indicate a serious eye condition such as acute angle glaucoma, iritis, keratitis or scleritis. Features such as significant pain, photophobia, reduced visual acuity and a unilateral presentation are “red flags” that a sight-threatening condition may be present. In the absence of specialised eye examination equipment, such as a slit lamp, General Practitioners must rely on identifying these key features to know which patients require referral to an Ophthalmologist for further assessment. Is it conjunctivitis or is it something more Iritis is also known as anterior uveitis; posterior uveitis is serious? inflammation of the choroid (choroiditis). Complications include glaucoma, cataract and macular oedema. The most likely cause of a red eye in patients who present to 4. Scleritis is inflammation of the sclera. This is a very rare general practice is conjunctivitis. However, red eye can also be presentation, usually associated with autoimmune a feature of a more serious eye condition, in which a delay in disease, e.g. rheumatoid arthritis. treatment due to a missed diagnosis can result in permanent 5. Penetrating eye injury or embedded foreign body; red visual loss. In addition, the inappropriate use of antibacterial eye is not always a feature topical eye preparations contributes to antimicrobial 6. Acid or alkali burn to the eye resistance. The patient history will usually identify a penetrating eye injury Most general practice clinics will not have access to specialised or chemical burn to the eye, but further assessment may be equipment for eye examination, e.g. -
UVEITIS Eye74 (1)
UVEITIS Eye74 (1) Uveitis Last updated: May 9, 2019 Classification .................................................................................................................................... 1 Etiologic categories .......................................................................................................................... 2 Treatment ......................................................................................................................................... 2 Complications ................................................................................................................................... 2 COMMON UVEITIC SYNDROMES ............................................................................................................. 2 Masquerade Syndromes ................................................................................................................... 3 UVEITIS - heterogenous ocular diseases - inflammation of any component of uveal tract (iris, ciliary body, choroid). CLASSIFICATION ANTERIOR UVEITIS (most common uveitis) - localized to anterior segment - iritis and iridocyclitis. IRITIS - white cells confined solely to anterior chamber. IRIDOCYCLITIS - cellular activity also involves retrolental vitreous. etiology (most do not have underlying systemic disease): 1) idiopathic postviral syndrome (most commonly 38-60%) 2) HLA-B27 syndromes, many arthritic syndromes (≈ 17%) 3) trauma (5.7%) 4) herpes simplex, herpes zoster disease (1.9-12.4%) 5) iatrogenic (postoperative). tends to -

View Returned the Following Day, Less Than OR
s s CONSEQUENTIAL CASES CASES THAT CHANGED THE WAY WE PRACTICE Three retina specialists share stories of patient encounters that left lasting impressions. LET THERE BE LIGHT By Ninel Z. Gregori, MD vision requires daily practice and hard my career in the direction of other A 52-year-old patient work. The patient had an intense innovative surgeries and clinical trials who had seen nothing rehabilitation process ahead of her at Bascom Palmer. but weak light for to help her to make sense of the And it did much more: It helped me 19 years since devel- lights. She is now able to identify light truly understand and appreciate the oping end-stage retinitis pigmentosa objects against dark backgrounds, value of careful preoperative counsel- (RP) came into my care. Then, in 2013, sort light and dark clothes, see light ing and setting realistic expectations the long-awaited promise of artificial coming in the windows, identify the for patients undergoing new treat- vision became a reality because the handle on the refrigerator, and even ments. After observing the experiences US FDA had approved the first retinal identify some letters, numbers, and of my patient receiving an Argus II prosthesis for patients with RP. I dis- simple words on a computer screen. implant, I now make sure to carefully cussed the Argus II Retinal Prosthesis She cannot, however, recognize explain the risks and limitations of System (Second Sight) with the objects and people in the environ- artificial vision options to patients who patient, and we agreed that she was a ment consistently, see faces, or read. -

Ocular Colobomaâ
Eye (2021) 35:2086–2109 https://doi.org/10.1038/s41433-021-01501-5 REVIEW ARTICLE Ocular coloboma—a comprehensive review for the clinician 1,2,3 4 5 5 6 1,2,3,7 Gopal Lingam ● Alok C. Sen ● Vijaya Lingam ● Muna Bhende ● Tapas Ranjan Padhi ● Su Xinyi Received: 7 November 2020 / Revised: 9 February 2021 / Accepted: 1 March 2021 / Published online: 21 March 2021 © The Author(s) 2021. This article is published with open access Abstract Typical ocular coloboma is caused by defective closure of the embryonal fissure. The occurrence of coloboma can be sporadic, hereditary (known or unknown gene defects) or associated with chromosomal abnormalities. Ocular colobomata are more often associated with systemic abnormalities when caused by chromosomal abnormalities. The ocular manifestations vary widely. At one extreme, the eye is hardly recognisable and non-functional—having been compressed by an orbital cyst, while at the other, one finds minimalistic involvement that hardly affects the structure and function of the eye. In the fundus, the variability involves the size of the coloboma (anteroposterior and transverse extent) and the involvement of the optic disc and fovea. The visual acuity is affected when coloboma involves disc and fovea, or is complicated by occurrence of retinal detachment, choroidal neovascular membrane, cataract, amblyopia due to uncorrected refractive errors, etc. While the basic birth anomaly cannot be corrected, most of the complications listed above are correctable to a great 1234567890();,: 1234567890();,: extent. Current day surgical management of coloboma-related retinal detachments has evolved to yield consistently good results. Cataract surgery in these eyes can pose a challenge due to a combination of microphthalmos and relatively hard lenses, resulting in increased risk of intra-operative complications. -

Ocular Findings Associated with Myelinated Retinal Nerve Fibers
Open Access Case Report DOI: 10.7759/cureus.14552 Ocular Findings Associated With Myelinated Retinal Nerve Fibers Jeslin Kera 1 , Airaj F. Fasiuddin 2 1. Ophthalmology, University of Central Florida College of Medicine, Orlando, USA 2. Ophthalmology, Nemours Children’s Hospital, Orlando, USA Corresponding author: Jeslin Kera, [email protected] Abstract The case involves a five-year-old female patient with a myelinated retinal nerve fiber (MRNF) layer of the right optic disc. Although this is a rare, benign, and often asymptomatic condition, it is sometimes associated with ocular findings which require early detection and treatment. In this case, the patient presented with strabismus, high myopia, and amblyopia. She was found to have myelinated retinal fiber layer lesions of the superotemporal and inferotemporal retina of her right eye. This case report aims to demonstrate the importance of performing a thorough evaluation of MRNF in the pediatric patient as well as to increase awareness of this entity to avoid misdiagnosis. Categories: Ophthalmology, Pediatrics Keywords: mrnf, myelinated retinal nerve fiber layer, optic nerve myelination, amblyopia, strabismus, high myopia, leukocoria, anisometropia Introduction Myelinated retinal nerve fiber (MRNF) layer is a rare and mostly benign congenital anomaly in which the retinal nerve fibers anterior to the lamina cribrosa have a myelin sheath. Normally, the optic nerve myelination does not extend past the lamina cribrosa and into the retina. Although the direct cause is unknown, MRNF occurs when the myelination extends past this point and is detectable on the fundus examination, obscuring the underlying retinal vessels. This anomaly can be present in up to 1% of the population, and approximately 7% of the affected patients will have bilateral involvement. -

Approach to Leukocoria Intro Hi Everyone, My Name Is
PedsCases Podcast Scripts This is a text version of a podcast from Pedscases.com on the “Approach to Leukocoria.” These podcasts are designed to give medical students an overview of key topics in pediatrics. The audio versions are accessible on iTunes or at www.pedcases.com/podcasts. Approach to Leukocoria Developed by Chris Novak, Dr. Natashka Pollock and Dr. Mel Lewis for PedsCases.com. March 1, 2016 Intro Hi everyone, my name is Chris Novak. I am a medical student at the University of Alberta. Today’s podcast is designed to give medical students an organized approach to the physical exam finding of leukocoria, or an abnormal red reflex in children. This podcast was developed with Dr. Melanie Lewis, a general pediatrician and Professor at the University of Alberta and Dr. Natashka Pollock, a pediatric ophthalmologist at the Stollery Children’s Hospital in Edmonton, Alberta. The term leukocoria refers to a “white pupil” when examining the eye. In some cases leukocoria may be grossly apparent, or may be noticed by parents in photographs, however it is often an incidental finding on physical exam. While leukocoria is not a common finding, it is a critical sign of vision- and potentially life-threatening conditions including cataracts, hemorrhage, and malignancy like retinoblastoma. Different causes of leukocoria can present from the neonatal period and throughout childhood hence,the American Academy of Pediatrics recommends that all neonates have their red reflex examined before discharge from hospital, and subsequently at all well-child visits. Thus, it is important that all physicians looking after children recognize the critical nature of this presentation so that it is not missed. -

Ocular Inflammation Associated with Systemic Infection
Byung Gil Moon, et al. • Ocular Inflammation Associated with Systemic Infection HMR Review Ocular Inflammation Associated with Systemic Infection Hanyang Med Rev 2016;36:192-202 http://dx.doi.org/10.7599/hmr.2016.36.3.192 pISSN 1738-429X eISSN 2234-4446 Byung Gil Moon, Joo Yong Lee Department of Ophthalmology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea Systemic infections that are caused by various types of pathogenic organisms can be spread Correspondence to: Joo Yong Lee Department of Ophthalmology, Asan to the eyes as well as to other solid organs. Bacteria, parasites, and viruses can invade the Medical Center, University of Ulsan eyes via the bloodstream. Despite advances in the diagnosis and treatment of systemic in- College of Medicine, 88 Olympic-ro fections, many patients still suffer from endogenous ocular infections; this is particularly 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-3976 due to an increase in the number of immunosuppressed patients such as those with hu- Fax: +82-2-470-6440 man immunodeficiency virus infection, those who have had organ transplantations, and E-mail: [email protected] those being administered systemic chemotherapeutic and immunomodulating agents, which may increase the chance of ocular involvement. In this review, we clinically evalu- Received 2 July 2016 Revised 21 July 2016 ated posterior segment manifestations in the eye caused by hematogenous penetration Accepted 27 July 2016 of systemic infections. We focused on the conditions that ophthalmologists encounter This is an Open Access article distributed under most often and that require cooperation with other medical specialists.