Oxidative-Phosphorylation Genes from the Mitochondrial and Nuclear
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Selective Induction of Apoptosis in HIV-Infected Macrophages
Selective Induction of Apoptosis in HIV-infected Macrophages by Simon Xin Min Dong A thesis submitted in partial fulfillment of the requirements for the Doctorate in Philosophy degree in Microbiology and Immunology Department of Biochemistry, Microbiology, and Immunology Faculty of Medicine University of Ottawa © Simon Xin Min Dong, Ottawa, Canada, 2020 Abstract The eradication of Human Immunodeficiency Virus (HIV) from infected patients is one of the major medical problems of our time, primarily due to HIV reservoir formation. Macrophages play important roles in HIV reservoir formation: once infected, they shield HIV against host anti-viral immune responses and anti-retroviral therapies, help viral spread and establish infection in anatomically protected sites. Thus, it is imperative to selectively induce the apoptosis of HIV-infected macrophages for a complete cure of this disease. I hypothesize that HIV infection dysregulates the expression of some specific genes, which is essential to the survival of infected host cells, and these genes can be targeted to selectively induce the apoptosis of HIV-infected macrophages. My objective is to identify the genes that can be targeted to eradicate HIV reservoir in macrophages, and to briefly elucidate the mechanism of cell death induced by targeting one of the identified genes. A four-step strategy was proposed to reach the goal. First, 90k shRNA lentivirus pool technology and microarray analysis were employed in a genome-wide screen of genes and 28 promising genes were found. Second, siRNA silencing was applied to validate these genes with 2 different HIV-1 viruses; as a result, 4 genes, Cox7a2, Znf484, Cdk2, and Cstf2t, were identified to be novel gene targets. -

Cell Development and Peripheral Survival Heme Exporter FLVCR Is
Heme Exporter FLVCR Is Required for T Cell Development and Peripheral Survival Mary Philip, Scott A. Funkhouser, Edison Y. Chiu, Susan R. Phelps, Jeffrey J. Delrow, James Cox, Pamela J. Fink and This information is current as Janis L. Abkowitz of September 28, 2021. J Immunol 2015; 194:1677-1685; Prepublished online 12 January 2015; doi: 10.4049/jimmunol.1402172 http://www.jimmunol.org/content/194/4/1677 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2015/01/09/jimmunol.140217 Material 2.DCSupplemental http://www.jimmunol.org/ References This article cites 44 articles, 11 of which you can access for free at: http://www.jimmunol.org/content/194/4/1677.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision by guest on September 28, 2021 • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2015 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology Heme Exporter FLVCR Is Required for T Cell Development and Peripheral Survival Mary Philip,*,† Scott A. -

Multi-Targeted Mechanisms Underlying the Endothelial Protective Effects of the Diabetic-Safe Sweetener Erythritol
Multi-Targeted Mechanisms Underlying the Endothelial Protective Effects of the Diabetic-Safe Sweetener Erythritol Danie¨lle M. P. H. J. Boesten1*., Alvin Berger2.¤, Peter de Cock3, Hua Dong4, Bruce D. Hammock4, Gertjan J. M. den Hartog1, Aalt Bast1 1 Department of Toxicology, Maastricht University, Maastricht, The Netherlands, 2 Global Food Research, Cargill, Wayzata, Minnesota, United States of America, 3 Cargill RandD Center Europe, Vilvoorde, Belgium, 4 Department of Entomology and UCD Comprehensive Cancer Center, University of California Davis, Davis, California, United States of America Abstract Diabetes is characterized by hyperglycemia and development of vascular pathology. Endothelial cell dysfunction is a starting point for pathogenesis of vascular complications in diabetes. We previously showed the polyol erythritol to be a hydroxyl radical scavenger preventing endothelial cell dysfunction onset in diabetic rats. To unravel mechanisms, other than scavenging of radicals, by which erythritol mediates this protective effect, we evaluated effects of erythritol in endothelial cells exposed to normal (7 mM) and high glucose (30 mM) or diabetic stressors (e.g. SIN-1) using targeted and transcriptomic approaches. This study demonstrates that erythritol (i.e. under non-diabetic conditions) has minimal effects on endothelial cells. However, under hyperglycemic conditions erythritol protected endothelial cells against cell death induced by diabetic stressors (i.e. high glucose and peroxynitrite). Also a number of harmful effects caused by high glucose, e.g. increased nitric oxide release, are reversed. Additionally, total transcriptome analysis indicated that biological processes which are differentially regulated due to high glucose are corrected by erythritol. We conclude that erythritol protects endothelial cells during high glucose conditions via effects on multiple targets. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Mitoxplorer, a Visual Data Mining Platform To
mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, Cecilia Garcia-Perez, José Villaveces, Salma Gamal, Giovanni Cardone, Fabiana Perocchi, et al. To cite this version: Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, et al.. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dy- namics and mutations. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkz1128. hal-02394433 HAL Id: hal-02394433 https://hal-amu.archives-ouvertes.fr/hal-02394433 Submitted on 4 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gkz1128 Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gkz1128/5651332 by Bibliothèque de l'université la Méditerranée user on 04 December 2019 mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim1,†, Prasanna Koti1,†, Adrien Bonnard2, Fabio Marchiano3, Milena Durrbaum¨ 1, Cecilia Garcia-Perez4, Jose Villaveces1, Salma Gamal1, Giovanni Cardone1, Fabiana Perocchi4, Zuzana Storchova1,5 and Bianca H. -

Structure of the Intact 14-Subunit Human Cytochrome C Oxidase
www.nature.com/cr www.cell-research.com ARTICLE Structure of the intact 14-subunit human cytochrome c oxidase Shuai Zong 1, Meng Wu1, Jinke Gu1, Tianya Liu1, Runyu Guo1 and Maojun Yang1,2 Respiration is one of the most basic features of living organisms, and the electron transport chain complexes are probably the most complicated protein system in mitochondria. Complex-IV is the terminal enzyme of the electron transport chain, existing either as randomly scattered complexes or as a component of supercomplexes. NDUFA4 was previously assumed as a subunit of complex-I, but recent biochemical data suggested it may be a subunit of complex-IV. However, no structural evidence supporting this notion was available till now. Here we obtained the 3.3 Å resolution structure of complex-IV derived from the human supercomplex I1III2IV1 and assigned the NDUFA4 subunit into complex-IV. Intriguingly, NDUFA4 lies exactly at the dimeric interface observed in previously reported crystal structures of complex-IV homodimer which would preclude complex- IV dimerization. Combining previous structural and biochemical data shown by us and other groups, we propose that the intact complex-IV is a monomer containing 14 subunits. Cell Research (2018) 28:1026–1034; https://doi.org/10.1038/s41422-018-0071-1 INTRODUCTION states under physiological conditions, either being assembled into Mitochondria are critical to many cellular activities. Respiration is supercomplexes or freely scattered on mitochondrial inner the central function of mitochondria, and is exquisitely regulated membrane.36 NDUFA4 was originally considered as a subunit of in multiple ways in response to varying cell conditions.1–3 The Complex-I37 but was proposed to belong to Complex-IV recently.38 assembly of respiratory chain complexes, Complex I–IV (CI, NADH: However, the precise location of NDUFA4 in CIV remains unknown, ubiquinone oxidoreductase; CII, succinate:ubiquinone oxidoreduc- and thus this notion still lacks structural support. -

Mouse Cox7a2 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Cox7a2 Knockout Project (CRISPR/Cas9) Objective: To create a Cox7a2 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Cox7a2 gene (NCBI Reference Sequence: NM_009945 ; Ensembl: ENSMUSG00000032330 ) is located on Mouse chromosome 9. 4 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 4 (Transcript: ENSMUST00000034881). Exon 1~4 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 1 starts from about 0.4% of the coding region. Exon 1~4 covers 100.0% of the coding region. The size of effective KO region: ~4106 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 Legends Exon of mouse Cox7a2 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of start codon is aligned with itself to determine if there are tandem repeats. Tandem repeats are found in the dot plot matrix. The gRNA site is selected outside of these tandem repeats. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of stop codon is aligned with itself to determine if there are tandem repeats. -

Rcf2 Revealed in Cryo-EM Structures of Hypoxic Isoforms of Mature Mitochondrial III-IV Supercomplexes
Rcf2 revealed in cryo-EM structures of hypoxic isoforms of mature mitochondrial III-IV supercomplexes Andrew M. Hartleya, Brigitte Meunierb, Nikos Pinotsisa, and Amandine Maréchala,c,1 aInstitute of Structural and Molecular Biology, Birkbeck College, WC1E 7HX London, United Kingdom; bInstitute for Integrative Biology of the Cell (I2BC), CNRS, Commissariat à l’Énergie Atomique et aux Énergies Alternatives, Université Paris-Saclay, 91198 Gif-sur-Yvette, France; and cInstitute of Structural and Molecular Biology, University College London, WC1E 6BT London, United Kingdom Edited by Harry B. Gray, California Institute of Technology, Pasadena, CA, and approved March 12, 2020 (received for review November 25, 2019) The organization of the mitochondrial electron transport chain formed by CI, III, and IV in varying stoichiometries (11–14) and I– proteins into supercomplexes (SCs) is now undisputed; however, III2 SCs (11, 14). Overall, they reveal a conserved arrangement of their assembly process, or the role of differential expression CI and CIII across species, including strong protein–protein inter- isoforms, remain to be determined. In Saccharomyces cerevisiae, actions. Slightly different orientationsofCIIIrelativetoCIwithin cytochrome c oxidase (CIV) forms SCs of varying stoichiometry the respirasome have been reported that have been linked to in- bc with cytochrome 1 (CIII). Recent studies have revealed, in stability of the SC during the purification procedure, although the normoxic growth conditions, an interface made exclusively by possibility of a subpopulation of respirasomes in which CI is in the Cox5A, the only yeast respiratory protein that exists as one of deactivated state could not be excluded (9, 14). However, when two isoforms depending on oxygen levels. -
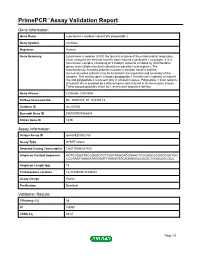
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name cytochrome c oxidase subunit VIa polypeptide 2 Gene Symbol COX6A2 Organism Human Gene Summary Cytochrome c oxidase (COX) the terminal enzyme of the mitochondrial respiratory chain catalyzes the electron transfer from reduced cytochrome c to oxygen. It is a heteromeric complex consisting of 3 catalytic subunits encoded by mitochondrial genes and multiple structural subunits encoded by nuclear genes. The mitochondrially-encoded subunits function in electron transfer and the nuclear-encoded subunits may be involved in the regulation and assembly of the complex. This nuclear gene encodes polypeptide 2 (heart/muscle isoform) of subunit VIa and polypeptide 2 is present only in striated muscles. Polypeptide 1 (liver isoform) of subunit VIa is encoded by a different gene and is found in all non-muscle tissues. These two polypeptides share 66% amino acid sequence identity. Gene Aliases COX6AH, COXVIAH RefSeq Accession No. NC_000016.9, NT_010393.16 UniGene ID Hs.250760 Ensembl Gene ID ENSG00000156885 Entrez Gene ID 1339 Assay Information Unique Assay ID qHsaCED0002180 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000287490 Amplicon Context Sequence GGTGCGGATGCGGAGGTGTTGGTAGGGACGGAACTCGGGGCGCGGGCGGTGG CCCGAGTGGAGATAGGAGTTGAAGGTGCAGAGGGCCACGCTGGGCAGCGCC Amplicon Length (bp) 73 Chromosome Location 16:31439339-31439441 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 98 R2 0.9997 cDNA Cq 28.87 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Role of Cytochrome C Oxidase Nuclear-Encoded Subunits in Health and Disease
Physiol. Res. 69: 947-965, 2020 https://doi.org/10.33549/physiolres.934446 REVIEW Role of Cytochrome c Oxidase Nuclear-Encoded Subunits in Health and Disease Kristýna ČUNÁTOVÁ1, David PAJUELO REGUERA1, Josef HOUŠTĚK1, Tomáš MRÁČEK1, Petr PECINA1 1Department of Bioenergetics, Institute of Physiology, Czech Academy of Sciences, Prague, Czech Republic Received February 2, 2020 Accepted September 13, 2020 Epub Ahead of Print November 2, 2020 Summary [email protected] and Tomáš Mráček, Department of Cytochrome c oxidase (COX), the terminal enzyme of Bioenergetics, Institute of Physiology CAS, Vídeňská 1083, 142 mitochondrial electron transport chain, couples electron transport 20 Prague 4, Czech Republic. E-mail: [email protected] to oxygen with generation of proton gradient indispensable for the production of vast majority of ATP molecules in mammalian Cytochrome c oxidase cells. The review summarizes current knowledge of COX structure and function of nuclear-encoded COX subunits, which may Energy demands of mammalian cells are mainly modulate enzyme activity according to various conditions. covered by ATP synthesis carried out by oxidative Moreover, some nuclear-encoded subunits possess tissue-specific phosphorylation apparatus (OXPHOS) located in the and development-specific isoforms, possibly enabling fine-tuning central bioenergetic organelle, mitochondria. OXPHOS is of COX function in individual tissues. The importance of nuclear- composed of five multi-subunit complexes embedded in encoded subunits is emphasized by recently discovered the inner mitochondrial membrane (IMM). Electron pathogenic mutations in patients with severe mitopathies. In transport from reduced substrates of complexes I and II to addition, proteins substoichiometrically associated with COX were cytochrome c oxidase (COX, complex IV, CIV) is found to contribute to COX activity regulation and stabilization of achieved by increasing redox potential of individual the respiratory supercomplexes.