Improving the Efficiency of Ammonia Electrolysis for Hydrogen Production
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Phosphine and Ammonia Photochemistry in Jupiter's
40th Lunar and Planetary Science Conference (2009) 1201.pdf PHOSPHINE AND AMMONIA PHOTOCHEMISTRY IN JUPITER’S TROPOSPHERE. C. Visscher1, A. D. Sperier2, J. I. Moses1, and T.C. Keane3. 1Lunar and Planetary Institute, USRA, 3600 Bay Area Blvd., Houston, TX 77058-1113 2Baylor University, Waco, TX 76798, 3Department of Chemistry and Physics, The Sage Colleges, Troy, NY 12180, ([email protected], [email protected]) Introduction: The last comprehensive photo- tions involving the amino radical (NH2) play a larger chemical model for Jupiter’s troposphere was pre- role. We note that this is the first photochemical sented by Edgington et al. [1,2], based upon the work model to include P2H as an intermediate species in PH3 of Atreya et al. [3] and Kaye and Strobel [4-6]. Since photolysis. The equivalent N2Hx species are believed these early studies, numerous laboratory experiments to be important in the combustion chemistry of nitro- have led to an improvement in our understanding of gen compounds [15]. PH3, NH3-PH3, and NH3-C2H2 photochemistry [7-13]. Furthermore, recent Galileo, Cassini, and Earth-based observations have better defined the abundance of key atmospheric constituents as a function of altitude and latitude in Jupiter’s troposphere. These new results provide an opportunity to test and improve theoretical models of Jovian atmospheric chemistry. We have therefore developed a photochemical model for Jupi- ter’s troposphere considering the updated experimental and observational constraints. Using the Caltech/JPL KINETICS code [14] for our photochemical models, our basic approach is two- fold. The validity of our selected chemical reaction list is first tested by simulating the laboratory experiments of PH3, NH3-PH3, and NH3-C2H2 photolysis with pho- tochemical “box” models. -

Ammonia As a Refrigerant
1791 Tullie Circle, NE. Atlanta, Georgia 30329-2305, USA www.ashrae.org ASHRAE Position Document on Ammonia as a Refrigerant Approved by ASHRAE Board of Directors February 1, 2017 Expires February 1, 2020 ASHRAE S H A P I N G T O M O R R O W ’ S B U I L T E N V I R O N M E N T T O D A Y © 2017 ASHRAE (www.ashrae.org). For personal use only. Additional reproduction, distribution, or transmission in either print or digital form is not permitted without ASHRAE’s prior written permission. COMMITTEE ROSTER The ASHRAE Position Document on “Ammonia as a Refrigerant” was developed by the Society’s Refrigeration Committee. Position Document Committee formed on January 8, 2016 with Dave Rule as its chair. Dave Rule, Chair Georgi Kazachki IIAR Dayton Phoenix Group Alexandria, VA, USA Dayton, OH, USA Ray Cole Richard Royal Axiom Engineers, Inc. Walmart Monterey, CA, USA Bentonville, Arkansas, USA Dan Dettmers Greg Scrivener IRC, University of Wisconsin Cold Dynamics Madison, WI, USA Meadow Lake, SK, Canada Derek Hamilton Azane Inc. San Francisco, CA, USA Other contributors: M. Kent Anderson Caleb Nelson Consultant Azane, Inc. Bethesda, MD, USA Missoula, MT, USA Cognizant Committees The chairperson of Refrigerant Committee also served as ex-officio members: Karim Amrane REF Committee AHRI Bethesda, MD, USA i © 2017 ASHRAE (www.ashrae.org). For personal use only. Additional reproduction, distribution, or transmission in either print or digital form is not permitted without ASHRAE’s prior written permission. HISTORY of REVISION / REAFFIRMATION / WITHDRAWAL -

Safer Cocktail for an Ethanol/Water/Ammonia Solvent
L A S C P LSC in Practice C P O C L Safer Cocktail for an Ethanol/Water/ K T I A I C Ammonia Solvent Mixture L S A Problem T A laboratory had been under pressure to move towards Using this mixture, the sample uptake capacity of I the newer generation of safer liquid scintillation ULTIMA-Flo AP, ULTIMA-Flo M and ULTIMA Gold O cocktails. Unfortunately, the sample composition of LLT (PerkinElmer 6013599, 6013579 and 6013377, one of their targets kept giving the researchers problems. respectively) at 20 °C was determined and N Before the lead researcher contacted PerkinElmer, the results were: they had tried PerkinElmer’s Opti-Fluor®, ULTIMA ULTIMA-Flo AP 4.00 mL in 10 mL cocktail N Gold LLT, and ULTIMA Gold XR. All of the safer O ™ ULTIMA-Flo M 4.25 mL in 10 mL cocktail cocktails could incorporate each of the constituents T as indicated by their sample capacity graphs. ULTIMA Gold LLT 4.25 mL in 10 mL cocktail E The problematic sample was an extraction solvent From these results, we observed that it was possible consisting of 900 mL ethanol, 50 mL ammonium to get 4.0 mL of the sample into all of these cocktails. hydroxide, 500 mL water and an enzyme containing In addition, we determined that it was not possible to 14C. The mixture had a pH in the range of 10 to 11. get 4 mL sample into 7 mL of any of these cocktails. To complete this work, we also checked for lumines- Discussion cence using 4 mL of sample in 10 mL of each cocktail. -

Cellulosic Ethanol Production Via Aqueous Ammonia Soaking Pretreatment and Simultaneous Saccharification and Fermentation Asli Isci Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2008 Cellulosic ethanol production via aqueous ammonia soaking pretreatment and simultaneous saccharification and fermentation Asli Isci Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Agriculture Commons, and the Bioresource and Agricultural Engineering Commons Recommended Citation Isci, Asli, "Cellulosic ethanol production via aqueous ammonia soaking pretreatment and simultaneous saccharification and fermentation" (2008). Retrospective Theses and Dissertations. 15695. https://lib.dr.iastate.edu/rtd/15695 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Cellulosic ethanol production via aqueous ammonia soaking pretreatment and simultaneous saccharification and fermentation by Asli Isci A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Co-majors: Agricultural and Biosystems Engineering; Biorenewable Resources and Technology Program of Study Committee: Robert P. Anex, Major Professor D. Raj Raman Anthony L. Pometto III. Kenneth J. Moore Robert C. Brown Iowa State University Ames, Iowa -

Algorithmic Analysis of Chemical Dynamics of the Autoignition of NH3–H2O2/Air Mixtures
energies Article Algorithmic Analysis of Chemical Dynamics of the Autoignition of NH3–H2O2/Air Mixtures Ahmed T. Khalil 1,2, Dimitris M. Manias 3, Efstathios-Al. Tingas 4, Dimitrios C. Kyritsis 1,2,* and Dimitris A. Goussis 1,2 1 Department of Mechanical Engineering, Khalifa University of Science and Technology, Abu Dhabi 127788, UAE; [email protected] (A.T.K.); [email protected] (D.A.G.) 2 Research and Innovation Center on CO2 and H2 (RICH), Khalifa University of Science and Technology, P.O. Box 127788, Abu Dhabi, UAE 3 Department of Mechanics, School of Applied Mathematics and Physical Sciences, National Technical University of Athens, 157 73 Athens, Greece; [email protected] 4 Perth College, University of the Highlands and Islands, (UHI), Perth PH1 2NX, UK; [email protected] * Correspondence: [email protected] Received: 8 September 2019; Accepted: 7 October 2019; Published: 21 November 2019 Abstract: The dynamics of a homogeneous adiabatic autoignition of an ammonia/air mixture at constant volume was studied, using the algorithmic tools of Computational Singular Perturbation. Since ammonia combustion is characterized by both unrealistically long ignition delays and elevated NOx emissions, the time frame of action of the modes that are responsible for ignition was analyzed by calculating the developing time scales throughout the process and by studying their possible relation to NOx emissions. The reactions that support or oppose the explosive time scale were identified, along with the variables that are related the most to the dynamics that drive the system to an explosion. -

BENZENE Disclaimer
United States Office of Air Quality EPA-454/R-98-011 Environmental Protection Planning And Standards June 1998 Agency Research Triangle Park, NC 27711 AIR EPA LOCATING AND ESTIMATING AIR EMISSIONS FROM SOURCES OF BENZENE Disclaimer This report has been reviewed by the Office of Air Quality Planning and Standards, U.S. Environmental Protection Agency, and has been approved for publication. Mention of trade names and commercial products does not constitute endorsement or recommendation of use. EPA-454/R-98-011 ii TABLE OF CONTENTS Section Page LIST OF TABLES.....................................................x LIST OF FIGURES.................................................. xvi EXECUTIVE SUMMARY.............................................xx 1.0 PURPOSE OF DOCUMENT .......................................... 1-1 2.0 OVERVIEW OF DOCUMENT CONTENTS.............................. 2-1 3.0 BACKGROUND INFORMATION ...................................... 3-1 3.1 NATURE OF POLLUTANT..................................... 3-1 3.2 OVERVIEW OF PRODUCTION AND USE ......................... 3-4 3.3 OVERVIEW OF EMISSIONS.................................... 3-8 4.0 EMISSIONS FROM BENZENE PRODUCTION ........................... 4-1 4.1 CATALYTIC REFORMING/SEPARATION PROCESS................ 4-7 4.1.1 Process Description for Catalytic Reforming/Separation........... 4-7 4.1.2 Benzene Emissions from Catalytic Reforming/Separation .......... 4-9 4.2 TOLUENE DEALKYLATION AND TOLUENE DISPROPORTIONATION PROCESS ............................ 4-11 4.2.1 Toluene Dealkylation -

Gas-Phase Reactions of Si+ with Ammonia and The
2396 J. Am. Chem. SOC.1988, 110, 2396-2399 Gas-Phase Reactions of Si+ with Ammonia and the Amines ( CH3)xNH3-x(x = 1-3): Possible Ion-Molecule Reaction Pathways toward SiH, SiCH, SiNH, SiCH3, SiNCH3, and H2SiNH S. Wlodek and D. K. Bohme* Contribution from the Department of Chemistry and Centre for Research in Experimental Space Science, York University, Downsview, Ontario, Canada M3J 1P3. Received August 31, 1987 Abstract: Rate constants and product distributions have been determined for gas-phase reactions of ground-state Si'(2P) ions with ammonia and the amines (CH,),NH3-, (x = 1-3) at 296 f 2 K with the selected-ion flow tube technique. All reactions were observed to be fast and can be understood in terms of Si' insertion into N-H and C-N bonds to form ions of the type SiNRIR2' (Rl, R2 = H, CHJ proceeding in competition (in the case of the amines) with hydride ion transfer to form immonium ions of the type CH2NRIR2' (Rl, R2 = H, CH,). C-N bond insertion appears more efficient than N-H bond insertion. The contribution of hydride ion transfer increases with increasing stability of the immonium ion. The latter reaction leads directly to SiH as a neutral product. Other minor reaction channels were seen which lead directly or indirectly to SiCH and SiCH3. Rapid secondary proton transfer reactions were observed for SiNH2' and SiNHCH3+to produce gas-phase SiNH and SiNCH3 molecules. With methylamine SiNH2+also appears to produce H2SiNH2' which may deprotonate to form the simplest silanimine, H2SiNH, or aminosilylene, HSiNH,. -

Ammonia/Ethanol Mixture for Adsorption Refrigeration
energies Article Ammonia/Ethanol Mixture for Adsorption Refrigeration Mauro Luberti, Chiara Di Santis and Giulio Santori * School of Engineering, Institute for Materials and Processes, the University of Edinburgh, Sanderson Building, King0s Buildings, Robert Stevenson Road, Edinburgh EH9 3FB, UK; [email protected] (M.L.); [email protected] (C.D.S.) * Correspondence: [email protected] Received: 29 January 2020; Accepted: 19 February 2020; Published: 22 February 2020 Abstract: Adsorption refrigeration has become an attractive technology due to the capability to exploit low-grade thermal energy for cooling power generation and the use of environmentally friendly refrigerants. Traditionally, these systems work with pure fluids such as water, ethanol, methanol, and ammonia. Nevertheless, the operating conditions make their commercialization still unfeasible, especially owing to safety and cost issues as a consequence of the working pressures, which are higher or lower than 1 atm. The present work represents the first thermodynamic insight in the use of mixtures for adsorption refrigeration and aims to assess the performance of a binary system of ammonia and ethanol. According to the Gibbs’ phase rule, the addition of a component introduces an additional degree of freedom, which allows to adjust the pressure of the system varying the composition of the mixture. The refrigeration process was simulated with isothermal- isochoric flash calculations to solve the phase equilibria, described by the Peng-Robinson-Stryjek-Vera (PRSV) equation of state for the vapor and liquid phases and by the ideal adsorbed solution theory (IAST) and the multicomponent potential theory of adsorption (MPTA) for the adsorbed phase. -

MATERIAL SAFETY DATA SHEET Ammonia, 2M Solution in Methanol
MATERIAL SAFETY DATA SHEET Ammonia, 2M solution in methanol Section 1 Chemical Product and Company Identification MSDS Name: Ammonia, 2M solution in methanol Catalog Numbers: 368390000, 368391000, 368398000 Synonyms: Company Identification: Acros Organics BVBA Janssen Pharmaceuticalaan 3a 2440 Geel, Belgium Company Identification: (USA) Acros Organics One Reagent Lane Fair Lawn, NJ 07410 For information in the US, call: 800ACROS01 For information in Europe, call: +32 14 57 52 11 Emergency Number, Europe: +32 14 57 52 99 Emergency Number US: 2017967100 CHEMTREC Phone Number, US: 8004249300 CHEMTREC Phone Number, Europe: 7035273887 Section 2 Composition, Information on Ingredients CAS# Chemical Name: % EINECS# Hazard Symbols: Risk Phrases: 67561 Methyl alcohol 96% 2006596 F T 11 23/24/25 39/23/24/25 7664417 Ammonia 4% 2316353 XN 10 20 36/37/38 Text for Rphrases: see Section 16 Hazard Symbols: T F Risk Phrases: 11 23/24/25 36/37/38 39/23/24/25 Section 3 Hazards Identification EMERGENCY OVERVIEW Highly flammable. Toxic by inhalation, in contact with skin and if swallowed. Irritating to eyes, respiratory system and skin. Toxic : danger of very serious irreversible effects through inhalation, in contact with skin and if swallowed. Potential Health Effects Eye: Produces irritation, characterized by a burning sensation, redness, tearing, inflammation, and possible corneal injury. May cause painful sensitization to light. Skin: Causes moderate skin irritation. May be absorbed through the skin in harmful amounts. Prolonged and/or repeated contact may cause defatting of the skin and dermatitis. -

Decarbonization of Industrial Sectors: the Next Frontier
Decarbonization of industrial sectors: the next frontier June 2018 Decarbonization of industrial sectors: the next frontier June 2018 Authored by: Arnout de Pee Dickon Pinner Occo Roelofsen Ken Somers Eveline Speelman Maaike Witteveen Preface The industrial sector is a vital source of wealth, prosperity, and social value on a global scale. Industrial companies produce about one-quarter of global GDP and employment, and make materials and goods that are integral to our daily lives, such as fertilizer to feed the growing global population, steel and plastics for the cars we drive, and cement for the buildings we live and work in. Industry also emits about 28 percent of global greenhouse gas (GHG) emissions, of which 90 percent are carbon dioxide (CO2) emissions. Between 1990 and 2014, GHG emissions from major sectors such as buildings, power, and transport increased by 23 percent (0.9 percent per year), while emissions from the industrial sector increased by 69 percent (2.2 percent per year). Over the last decades, the outlines of energy transition pathways have emerged in the buildings, power and transport sectors. These have been driven by technological breakthroughs and cost reduction. For industrial processes, such pathways are less well-defined. The energy transition in industry should be viewed in the context of global trends that will impact the demand for and preferred production routes of industrial products. There is an expected growth in resource demand, driven by an increase in middle class consumers of ~3 billion in the coming 20 years, as well as rapid urbanization. This coincides with growing constraints on key resources, such as copper and zinc, and environmental degradation, for instance from air pollution. -

Ammonia-Nitrogen (NH3-N) and Ammonium-Nitrogen (NH4 -N) Equilibrium on the Process of Removing Nitrogen by Using Tubular Plastic Media
Journal(of(Materials(and(( J. Mater. Environ. Sci., 2017, Volume 8, Issue S, Page 4915-4922 Environmental(Sciences( ISSN(:(2028;2508( CODEN(:(JMESCN( http://www.jmaterenvironsci.com ! Copyright(©(2017,((((((((((((((((((((((((((((( University(of(Mohammed(Premier(((((( OuJda(Morocco( + Ammonia-Nitrogen (NH3-N) and Ammonium-Nitrogen (NH4 -N) Equilibrium on The Process of Removing Nitrogen By Using Tubular Plastic Media Purwono, Arya Rezagama, Muhammad Hibbaan, Mochamad Arief Budihardjo Environmental Engineering Department, Faculty of Engineering, Diponegoro University, Semarang, Indonesia Received 08 Oct 2016, Abstract Revised 09 Nov 2017, The biological process of nitrification-denitrification, involving a tubular plastic bio + Accepted 17 Nov 2017 filter, reduces nitrogen in wastewater. During the process, ionised ammonia (NH4 ) and unionised ammonia (NH3) are formed simultaneously in the water. The presence + Keywords of NH4 causes the breakpoint of chlorination and the formation of disinfection by- !!unionized ammonia products (DBPs) in the form of trihalomethana, while unionised ammonia (NH3) is !!ionized ammonia toxic to aquatic organisms. In this study, total ammonia nitrogen (TAN) removal !!aerobic biofilter efficiency using a tubular plastic bio filter was 14.31% with pH of 7.89 on the first !!nitrogen removal day. TAN concentration in the bio filter reactor was 77.08 mg/l, and the NH3-N + !!equilibrium equilibrium reached 4.8 mg of NH3-N/l while NH4 -N equilibrium went to 72.2 mg of + NH4 -N/l. In the control reactor, TAN removal efficiency was 12.98%, where the [email protected] + NH3-N equilibrium touched 1.94 mg of NH3-N/l, and the NH4 equilibrium reached Phone: +62 24 76480678; + 76.33 mg of NH4 -N/l with a pH of 7.56. -
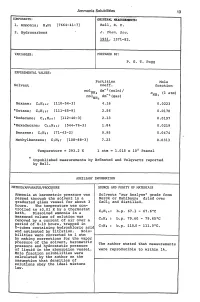
Ammonia Solubilities 13 VARIABLES: PREPARED BY: EXPERIMENTAL
Ammonia Solubilities 13 COHPONENTS: ORIGINAL MEASUREMENTS: 1. Anunonia; HsN; [7664-41-7] Bell, R. P. 2. Hydrocarbons J. Chern. Soa. 1931, 1371-82. VARIABLES: PREPARED BY: P. G. T. Fogg EXPERIMENTAL VALUES: Partition Mole Solvent coeff. fraction mo1 dm- s (soln)/ NHs X (1 atm) rno1 dm-s(gas) NHs NHs Hexane; C6H14; [110-54-3] 4.16 0.0223 *Octane; CSH1S; [111-65-9] 2.56 0.0170 *Dodecane; C12H26; [112-40-3] 2.13 0.0197 *Hexadecane; C16HS4; [544-76-3] 1. 84 0.0219 Benzene; C6H6; [71-43-2] 9.95 0.0474 Methy1benzene; C7HS; [108-88-3] 7.23 0.0313 Temperature = 293.2 K 1 atm = 1.015 x 10 5 Pascal * Unpublished measurements by Bronsted and Vo1qvartz reported by Bell. AUXILIARY INFORMATION METHOD/APPARATUS/PROCEDURE SOURCE AND PURITY OF MATERIALS \ Ammonia at barometric pressure was Solvents "zur Analyse" grade from passed through the solvent in a Merck or Kah1baum; dried over graduated glass vessel for about 3 CaC12 and distilled. hours. The temperature was cpn trolled to to.01 K by a thermostat bath. Dissolved ammonia in a C6H14: b.p. 67.1 - 67.6°C measured volume of solution was removed by a current of air over a C6H6: b.p. 79.60 - 79.65°C period of 8-10 hours, trapped in C7HS: b.p. 110.0 - 111. ooC. U-tubes containing hydrochloric acic and estimated by titration. Solu- bilities were corrected to 1 atm 1---------------------1 by making corrections for the vapor pressure of the solvent, barometric The author stated that measurements pressure and hydrostatic pressure of liquid in the absorption vessel.