Group 3 and Group 13 Metal Hydride Compounds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

LIGHT METAL BOROHYDRIDES and Mg-BASED HYDRIDES for HYDROGEN STORAGE
LIGHT METAL BOROHYDRIDES AND Mg-BASED HYDRIDES FOR HYDROGEN STORAGE by SHENG GUO A thesis submitted to the University of Birmingham for the degree of DOCTOR OF PHILOSOPHY School of Metallurgy and Materials College of Engineering and Physical Sciences University of Birmingham December 2014 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. Synopsis This work has investigated structural and compositional changes in LiBH4, Mg(BH4)2, Ca(BH4)2, LiBH4-Ca(BH4)2 during heating. The crystal and vibrational structures of these borohydrides/composites were characterized using lab-based X-ray diffraction (XRD) and Raman spectroscopy, with particular attention to the frequency/width changes of Raman vibrations of different polymorphs of borohydrides. The thermal stability and decomposition pathway of the borohydrides was studied in great detail mainly using differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), in/ex situ XRD and Raman measurements, whilst the gaseous products during heating were monitored using a mass spectrometry (MS). Hydrogen is the main decomposition gaseous product from all of these compounds, but in some cases a very small amount of diborane release was also detected. -

NNL TSL Evaluations for Yttrium Hydride and Hexagonal Ice
NNL TSL Evaluations for Yttrium Hydride and Hexagonal Ice Michael Zerkle and Jesse Holmes Naval Nuclear Laboratory Cross Section Evaluation Working Group Meeting November 14-16, 2016 Brookhaven National Laboratory The Naval Nuclear Laboratory is operated for the U.S. Department of Energy and the U.S. Department of the Navy by Bechtel Marine Propulsion Corporation, a wholly owned subsidiary of Bechtel National, Inc. NNL TSL Evaluations Material MAT H(YH2) 5 Y(YH2) 55 H(ice-Ih) 10 O(ice-Ih) 50 2 YH2 Background • Yttrium hydride (YH2) is an advanced high temperature moderator • Superior hydrogen density (NH) at elevated temperatures • Keinert (1971) proposed H(YH2) TSL based on simple analytic frequency distributions • Debye-type for acoustic mode • Gaussian-type for optical mode • Higher-fidelity TSLs for H(YH2) and Y(YH2) developed using first- principles calculations • Density Functional Theory (DFT) to calculate interatomic Hellman- Feynman forces for crystal structure Hydrogen in metallic Zr, Ce, Y and Ca in equilibrium with 1 atm H at various temperatures. • Lattice Dynamics (LD) to determine 2 dispersion relations and phonon (Source: Metal Hydrides, Academic Press, p. 442, 1968) density of states (DOS) -22 NH is number of hydrogen atoms/cc x 10 3 Ab Initio Calculation of Phonon Spectra 4 YH2 Structure and Lattice Dynamics • YH2 has a CaF2 type FCC structure • 12 atoms • 4 Y atoms (blue) at vertices and faces of unit cell • 8 H atoms (grey) in tetrahedral holes between Y atoms • Dispersion relations (at right) • Well separated acoustical and optical modes • Lower branches are acoustical modes mainly due to heavy Y atom vibrations • Higher branches are optical modes mainly due to light H atom vibrations 5 YH2 Phonon Density of States 6 YH2 Phonon Density of States High resolution inelastic neutron scattering spectra from Udovic shows similar structure for YH2 centered on 0.127 eV. -
Chapter 13.Pptx
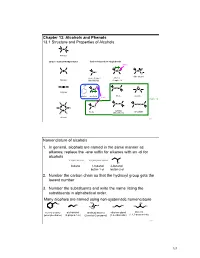
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

NETS 2020 Template
بÀƵƧǘȁǞƧƊǶ §ȲȌǐȲƊǿ ƊƧDzɈȌɈǘƵwȌȌȁƊȁƮȌȁ ɈȌwƊȲȺɈǘȲȌɐǐǘƊƮɨƊȁƧǞȁǐ خȁɐƧǶƵƊȲɈƵƧǘȁȌǶȌǐǞƵȺƊȁƮ ǞȁȁȌɨƊɈǞȌȁ ǞȺ ȺȯȌȁȺȌȲƵƮ Ʀɯ ɈǘƵ ƊDz ªǞƮǐƵ yƊɈǞȌȁƊǶ ׁׂ׀ׂ y0À² ÀǘǞȺ ƧȌȁǏƵȲƵȁƧƵ خׁׂ׀ׂ ةɈǘ׀׃ƊȁƮ ɩǞǶǶƦƵ ǘƵǶƮ ǏȲȌǿȯȲǞǶ ׂ׆ɈǘٌةmƊƦȌȲƊɈȌȲɯ ɩǞǶǶ ƦƵ ǘƵǶƮ ɨǞȲɈɐƊǶǶɯ ȺȌ ɈǘƊɈ ɈǘƵ ƵȁɈǞȲƵ y0À² خƧȌǿǿɐȁǞɈɯǿƊɯȯƊȲɈǞƧǞȯƊɈƵǞȁɈǘǞȺƵɮƧǞɈǞȁǐǿƵƵɈǞȁǐ ǐȌɨخȌȲȁǶخخׁׂ׀ȁƵɈȺׂششبǘɈɈȯȺ Nuclear and Emerging Technologies for Space Sponsored by Oak Ridge National Laboratory, April 26th-30th, 2021. Available online at https://nets2021.ornl.gov Table of Contents Table of Contents .................................................................................................................................................... 1 Thanks to the NETS2021 Sponsors! ...................................................................................................................... 2 Nuclear and Emerging Technologies for Space 2021 – Schedule at a Glance ................................................. 3 Nuclear and Emerging Technologies for Space 2021 – Technical Sessions and Panels By Track ............... 6 Nuclear and Emerging Technologies for Space 2021 – Lightning Talk Final Program ................................... 8 Nuclear and Emerging Technologies for Space 2021 – Track 1 Final Program ............................................. 11 Nuclear and Emerging Technologies for Space 2021 – Track 2 Final Program ............................................. 14 Nuclear and Emerging Technologies for Space 2021 – Track 3 Final Program ............................................. 18 -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

Catalytic Systems Based on Cp2zrx2 (X = Cl, H), Organoaluminum
catalysts Article Catalytic Systems Based on Cp2ZrX2 (X = Cl, H), Organoaluminum Compounds and Perfluorophenylboranes: Role of Zr,Zr- and Zr,Al-Hydride Intermediates in Alkene Dimerization and Oligomerization Lyudmila V. Parfenova 1,* , Pavel V. Kovyazin 1, Almira Kh. Bikmeeva 1 and Eldar R. Palatov 2 1 Institute of Petrochemistry and Catalysis of Russian Academy of Sciences, Prospekt Oktyabrya, 141, 450075 Ufa, Russia; [email protected] (P.V.K.); [email protected] (A.K.B.) 2 Bashkir State University, st. Zaki Validi, 32, 450076 Ufa, Russia; [email protected] * Correspondence: [email protected]; Tel.: +7-347-284-3527 i i Abstract: The activity and chemoselectivity of the Cp2ZrCl2-XAlBu 2 (X = H, Bu ) and [Cp2ZrH2]2- ClAlEt2 catalytic systems activated by (Ph3C)[B(C6F5)4] or B(C6F5)3 were studied in reactions with 1-hexene. The activation of the systems by B(C6F5)3 resulted in the selective formation of head- to-tail alkene dimers in up to 93% yields. NMR studies of the reactions of Zr complexes with organoaluminum compounds (OACs) and boron activators showed the formation of Zr,Zr- and Zr,Al-hydride intermediates, for which diffusion coefficients, hydrodynamic radii, and volumes were estimated using the diffusion ordered spectroscopy DOSY. Bis-zirconium hydride clusters of type x[Cp ZrH ·Cp ZrHCl·ClAlR ]·yRnAl(C F ) − were found to be the key intermediates of alkene 2 2 2 2 6 5 3 n dimerization, whereas cationic Zr,Al-hydrides led to the formation of oligomers. Citation: Parfenova, L.V.; Kovyazin, P.V.; Bikmeeva, A.K.; Palatov, E.R. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive. -

Hydrogen Transmutation of Nickel in Glow Discharge
International Journal of Materials Science ISSN 0973-4589 Volume 12, Number 3 (2017), pp. 405-409 © Research India Publications http://www.ripublication.com Hydrogen Transmutation of Nickel in Glow Discharge Vladimir K. Nevolin National Research University of Electronic Technology (MIET), Moscow, Russia. Abstract Background: The possibility of the existence of subatomic hydrogen states was theoretically predicted previously. Objectives: Prove that the transmutation of elements is possible in specially prepared conditions for hydrogen. Methodology: By comparing the mass spectra of deposits on silicon substrates and target electrodes, it is shown that a change in the composition is observed in a magnetron Argon. Results: An increase in the concentration of 62 60 the nickel isotope 28 Ni and a decrease in the isotope concentration 28 Ni are shown. Conclusion: These results confirm the results obtained earlier in the heat generator Rossi, who worked more than a year, found an increase in the 62 isotope 28 Ni due to a decrease in the proportion of other isotopes. Keywords: transmutation, isotopes of nickel, glow discharge, argon, hydrogen INTRODUCION It is considered that the cold transmutation of elements (cold nuclear reactions) has been experimentally demonstrated [1]. On the basis of this phenomenon, energy generators are created in which long-term release of thermal energy in excess of expended energy is observed [2]. From many experimental studies it can be seen that hydrogen, which plays a pivotal role in the reaction zone, may be delivered through a variety of chemical compounds; for example, using lithium aluminium hydride LiAlH4. An analysis of the products of nuclear reactions suggests the possibility of many simultaneous nuclear fusion and decomposition reactions [3]. -

Synthesis, Reactivity, and Catalysis of Group 3 and Lanthanide Alkyl Complexes
Synthesis, Reactivity, and Catalysis of Group 3 and Lanthanide Alkyl Complexes By Daniel Steven Levine A dissertation submitted in partial satisfaction of the requirements for the degree of Doctor of Philosophy in Chemistry in the Graduate Division of the University of California, Berkeley Committee in charge: Professor T. Don Tilley, Co-Chair Professor Richard A. Andersen, Co-Chair Professor Alexis T. Bell Summer 2016 Abstract Synthesis, Reactivity, and Catalysis of Group 3 and Lanthanide Alkyl Complexes by Daniel Steven Levine Doctor of Philosophy in Chemistry University of California, Berkeley Professor T. Don Tilley, Co-Chair Professor Richard A. Andersen, Co-Chair Chapter 1. A series of scandium dialkyl complexes, (PNP)ScR2 (R = neopentyl, trimethylsilylmethyl), supported by the monoanionic, chelating PNP ligand (2,5- bis(dialkylphosphinomethyl)pyrrolide; alkyl = cyclohexyl, tert-butyl) was synthesized and the reactivities of these complexes toward simple hydrocarbons was investigated. The scandium– carbon bonds undergo σ-bond metathesis reactions with hydrogen and these complexes are catalysts for the hydrogenation of alkenes. Reactions with primary amines led to formation of amido complexes that undergo cyclometalation via σ-bond metathesis, without involvement of an imido complex intermediate. A variety of carbon-hydrogen bonds are also activated, including sp-, sp2-, and sp3-C–H bonds (intramolecularly in the latter case). Levine, D. S.; Tilley, T. D.; Andersen, R. A. Organometallics 2015, 34 (19), 4647. Chapter 2. Terminal group 3 methylidene complexes are generated by thermolysis of monoanionic PNP-supported scandium and yttrium dialkyl complexes. The reaction mechanism has been probed by deuterium-labeling experiments and DFT calculations. Abstraction of a γ- hydrogen from one alkyl group by the other affords a metallacyclobutane that undergoes [2+2] cycloreversion, analogous to a key step in the olefin metathesis reaction, to generate a methylidene complex and isobutene. -

WO 2015/177807 Al 26 November 2015 (26.11.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/177807 Al 26 November 2015 (26.11.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 403/14 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/IN20 15/000066 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 3 February 2015 (03.02.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 1709/MUM/2014 22 May 2014 (22.05.2014) (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: WANBURY LTD. [IN/IN]; BSEL tech park, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, B wing, 10th floor, sector 30A, opp. -

Transition Metal Hydrides
Transition Metal Hydrides Biomimetic Studies and Catalytic Applications Jesper Ekström Stockholm University © Jesper Ekström, Stockholm 2007 ISBN 978-91-7155-539-7 Printed in Sweden by US-AB, Stockholm 2007 Distributor: Department of Organic Chemistry ‘Var som en anka brukade min mamma alltid säga. Håll dig lugn på ytan, och paddla utav bara helvete därunder.’ Michael Caine Abstract In this thesis, studies of the nature of different transition metal-hydride com- plexes are described. The first part deals with the enantioswitchable behav- iour of rhodium complexes derived from amino acids, applied in asymmetric transfer hydrogenation of ketones. We found that the use of amino acid thio amide ligands resulted in the formation of the R-configured product, whereas the use of the corresponding hydroxamic acid- or hydrazide ligands selec- tively gave the S-alcohol. Structure/activity investigations revealed that the stereochemical outcome of the catalytic reaction depends on the ligand mode of coordination. In the second part, an Fe hydrogenase active site model complex with a la- bile amine ligand has been synthesized and studied. The aim of this study was to find a complex that efficiently catalyzes the reduction of protons to molecular hydrogen under mild conditions. We found that the amine ligand functions as a mimic of the loosely bound ligand which is part of the active site in the hydrogenase. Further, an Fe hydrogenase active site model complex has been coupled to a photosensitizer with the aim of achieving light induced hydrogen production. The redox properties of the produced complex are such that no electron transfer from the photosensitizer part to the Fe moiety occurs.