Investigating Neural Stem and Progenitor Cell Intracrine Signaling Presented in Partial Fulfilment of the Requirements the Maste
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Intracrine View of Sex Steroids, Immunity, and Metabolic Regulation
Review An intracrine view of sex steroids, immunity, and metabolic regulation Katya B. Rubinow ABSTRACT Background: Over the past two decades, parallel recognition has grown of the importance of both sex steroids and immune activity in metabolic regulation. More recently, these discrete areas have been integrated in studies examining the metabolic effects of sex steroid immunomodulation. Implicit in these studies has been a traditional, endocrine model of sex steroid delivery from the gonads to target cells, including immune cells. Thus, research to date has focused on the metabolic effects of sex steroid receptor signaling in immune cells. This endocrine model, however, overlooks the extensive capacity of immune cells to generate and metabolize sex steroids, enabling the production of sex steroids for intracrine signaling e that is, sex steroid production for signaling within the cell of origin. Intracrine function allows highly cell-autonomous regulation of sex steroid exposure, and sex steroid secretion by immune cells could confer paracrine signaling effects in neighboring cells within metabolic tissues. In this review, immune cell intracrinology will denote sex steroid production within immune cells for either intracrine or paracrine signaling. This intracrine capacity of immune cells has been well established, and prior work has supported its importance in autoimmune disorders, trauma, and cancer. The potential relevance of immune cell intracrine function to the regulation of energy balance, body weight, body composition, and insulin sensitivity has yet to be explored. Scope of review: The following review will detail findings to date regarding the steroidogenic and steroid metabolizing capacity of immune cells, the regulation of immune cell intracrine function, and the biological effects of immune-derived sex steroids, including the clinical relevance of immune cell intracrinology in fields other than metabolism. -

Sulfation Through the Looking Glass—Recent Advances in Sulfotransferase Research for the Curious
The Pharmacogenomics Journal (2002) 2, 297–308 & 2002 Nature Publishing Group All rights reserved 1470-269X/02 $25.00 www.nature.com/tpj REVIEW Sulfation through the looking glass—recent advances in sulfotransferase research for the curious MWH Coughtrie ABSTRACT Members of the cytosolic sulfotransferase (SULT) superfamily catalyse the Department of Molecular & Cellular Pathology, sulfation of a multitude of xenobiotics, hormones and neurotransmitters. University of Dundee, Ninewells Hospital & Humans have at least 10 functional SULT genes, and a number of recent Medical School, Dundee, Scotland, UK advances reviewed here have furthered our understanding of SULT function. Correspondence: Analysis of expression patterns has shown that sulfotransferases are highly MWH Coughtrie, Department of expressed in the fetus, and SULTs may in fact be a major detoxification Molecular & Cellular Pathology, University enzyme system in the developing human. The X-ray crystal structures of of Dundee, Ninewells Hospital and three SULTs have been solved and combined with mutagenesis experiments Medical School, Dundee DD1 9SY, Scotland, UK. and molecular modelling, they have provided the first clues as to the factors Tel: +44 (0)1382 632510 that govern the unique substrate specificities of some of these enzymes. In Fax: +44 (0)1382 640320 the future these and other studies will facilitate prediction of the fate of E-mail: [email protected] chemicals metabolised by sulfation. Variation in sulfation capacity may be important in determining an individual’s response to xenobiotics, and there has been an explosion in information on sulfotransferase polymorphisms and their functional consequences, including the influence of SULT1A1 genotype on susceptibility to colorectal and breast cancer. -
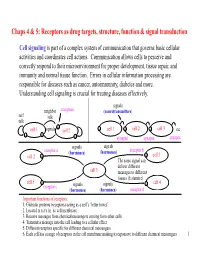
Chaps 4 & 5: Receptors As Drug Targets, Structure, Function & Signal Transduction
Chaps 4 & 5: Receptors as drug targets, structure, function & signal transduction Cell signaling is part of a complex system of communication that governs basic cellular activities and coordinates cell actions. Communication allows cells to perceive and correctly respond to their microenvironment for proper development, tissue repair, and immunity and normal tissue function. Errors in cellular information processing are responsible for diseases such as cancer, autoimmunity, diabetes and more. Understanding cell signaling is crucial for treating diseases effectively. signals neighbor receptors (neurotransmitters) self talk talk cell 1 signals cell 2 cell 1 cell 2 cell 3 etc. synapse synapse synapse signals signals receptor a (hormones) (hormones) receptor b cell 2 cell 3 The same signal can deliver different cell 1 messages to different tissues (histamine). cell 5 cell 4 receptor c signals signals (hormones) (hormones) receptor d Important functions of receptors: 1. Globular proteins (receptors) acting as a cell’s ‘letter boxes’ 2. Located mostly in the cell membrane 3. Receive messages from chemical messengers coming from other cells 4. Transmit a message into the cell leading to a cellular effect 5. Different receptors specific for different chemical messengers 6. Each cell has a range of receptors in the cell membrane making it responsive to different chemical messengers 1 © Oxford University Press, 2013 Signals can be divided into the 5 catagories below. Signal carriers (S) have to reach the proper receptors (R) for the message to be recieved. Intracrine signals are produced by the target cell that stay within the target cell. cell a S = signal S R R = receptor Autocrine signals are produced by the target cell, are secreted, and affect the target cell itself via receptors. -

Cell Plasticity and Prostate Cancer: the Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance
cancers Review Cell Plasticity and Prostate Cancer: The Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance Sofia Papanikolaou, Aikaterini Vourda, Spyros Syggelos and Kostis Gyftopoulos * Department of Anatomy, University of Patras School of Medicine, Rion, 26504 Patras, Greece; [email protected] (S.P.); [email protected] (A.V.); [email protected] (S.S.) * Correspondence: [email protected] Simple Summary: Although epithelial-to-mesenchymal transition (EMT) is a well-known cellular process involved during normal embryogenesis and wound healing, it also has a dark side; it is a complex process that provides tumor cells with a more aggressive phenotype, facilitating tumor metastasis and even resistance to therapy. This review focuses on the key pathways of EMT in the pathogenesis of prostate cancer and the development of metastases and evasion of currently available treatments. Abstract: Prostate cancer, the second most common malignancy in men, is characterized by high heterogeneity that poses several therapeutic challenges. Epithelial–mesenchymal transition (EMT) is a dynamic, reversible cellular process which is essential in normal embryonic morphogenesis and Citation: Papanikolaou, S.; Vourda, wound healing. However, the cellular changes that are induced by EMT suggest that it may also A.; Syggelos, S.; Gyftopoulos, K. Cell play a central role in tumor progression, invasion, metastasis, and resistance to current therapeutic Plasticity and Prostate Cancer: The options. These changes include enhanced motility and loss of cell–cell adhesion that form a more Role of Epithelial–Mesenchymal aggressive cellular phenotype. Moreover, the reverse process (MET) is a necessary element of the Transition in Tumor Progression, Invasion, Metastasis and Cancer metastatic tumor process. -

The Enigmatic Processing and Secretion of Interleukin-33
Cellular & Molecular Immunology (2010) 7, 260–262 ß 2010 CSI and USTC. All rights reserved 1672-7681/10 $32.00 www.nature.com/cmi MINI REVIEW The enigmatic processing and secretion of interleukin-33 Weihua Zhao and Zhiqing Hu Interleukin-33 (IL-33) is the most attractive novel cytokine identified as an IL-1 family member. IL-33 was first named NF-HEV (nuclear factor from high endothelial venules), as it was known to interact with nuclear chromatin although its exact intracellular functions are still to be clarified. IL-33 is now recognized as the specific ligand for the orphan IL-1 receptor family member ST2 and to be involved in polarization of T cells towards T helper 2 cell phenotype and in activation of mast cells, bosophils, eosinophils and natural killer cells. It is essential for IL-33 to be extracellularly released in order to bind to the ST2 receptor and consequently play a crucial role in inflammatory, infectious and autoimmune diseases. However, like the IL-1 family members, IL-1b and IL-18, IL-33 mRNA is translated without a signal sequence for secretion. Additionally, IL-33 cannot be released by the processing and secretion mechanism shared by IL-1b and IL-18 as IL-33 is not a substrate of caspase-1 and does not require proteolysis for activation. In contrast, IL-33 can be inactivated by apoptotic caspases. Accordingly, IL-33 is proposed to be released as an alarmin from necrotic cells but to be deleted during apoptosis. Besides the known autocrine, paracrine, intracrine, juxtacrine and retrocrine mechanisms of cellular interaction with cytokines, release by necrotic cells is another pathway for a cytokine to display its function, which we suggest might be called ‘necrocrine’. -

Thirty Years of Intracrinology
The Ochsner Journal 14:673–680, 2014 Ó Academic Division of Ochsner Clinic Foundation Thirty Years of Intracrinology Richard N. Re, MD Research Division, Ochsner Health System, New Orleans, LA lular signaling molecules and could also act in the ABSTRACT intracellular space: these varied factors displayed Background: Intracrinology is the study of the intracellular intracrine functionality. Surprisingly, included among actions, regulation, trafficking, and interactions of extracellular these proteins were growth factors, cytokines, tran- signaling peptides/proteins. scription factors, DNA binding proteins, and en- 1-9 Methods: We describe the development of intracrine biology zymes. since the term was defined in 1984. We developed the basic principles of intracrine action based on observing the functionality of various Results: Intracrine biology plays a role in many normal and intracrines.10-43 For example, many intracrines upre- pathological processes and represents a fertile field for the gulate either their own synthesis or the synthesis of development of novel therapeutics. elements of their signaling cascades. Intracrines can Conclusion: Although 30 years old, the field of intracrinology is operate in positive feedback loops—that is, in what only now becoming widely accepted. Intracrine principles can has been termed a feed-forward mode. For example, be applied to the investigation of physiological processes and angiotensin II action at the nucleus can upregulate the to the development of new therapies. transcription of renin and angiotensinogen. Investiga- tors have reported that high glucose levels stimulate the synthesis of intracellular angiotensin II and establish a feed-forward loop in which angiotensin II INTRODUCTION upregulates angiotensinogen.35,38 This feed-forward In 1984, this laboratory introduced the term loop appears to play a role in diabetes-related intracrine, meaning the action of a peptide hormone pathology and in particular diabetic cardiomyopathy. -

The Precursor Form of IL-1 Is an Intracrine Proinflammatory Activator
The precursor form of IL-1␣ is an intracrine proinflammatory activator of transcription Ariel Werman*†, Rachel Werman-Venkert*, Rosalyn White*, Jae-Kwon Lee†, Batsheva Werman†, Yakov Krelin*, Elena Voronov*, Charles A. Dinarello†, and Ron N. Apte*‡ *Department of Microbiology and Immunology, Faculty of Health Sciences and Cancer Research Center, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israel; and †Department of Medicine, University of Colorado Health Sciences Center, Denver, CO 80262 Contributed by Charles A. Dinarello, December 31, 2003 Although most cytokines are studied for biological effects after additional external stimulus. For example, mice deficient in engagement of their specific cell surface membrane receptors, IL-1␣ are resistant to experimental colitis (unpublished obser- increasing evidence suggests that some function in the nucleus. In vations), and IL-1␣-deficient but not IL-1-deficient mice fail to the present study, the precursor form of IL-1␣ was overexpressed prime antigen-specific T cells (9). in various cells and assessed for activity in the presence of satu- The possibility that, upon nuclear localization sequence- rating concentrations of IL-1 receptor antagonist to prevent recep- dependent nuclear translocation, pIL-1␣ and propiece IL-1␣ tor signaling. Initially diffusely present in the cytoplasm of resting (ppIL-1␣) might exhibit functionality has been the focus of cells, IL-1␣ translocated to the to nucleus after activation by several studies. Indeed, overexpression of the propiece results in endotoxin, a Toll-like receptor ligand. The IL-1␣ precursor, but not transformation to a malignant phenotype (10). The propiece also the C-terminal mature form, activated the transcriptional machin- has been shown to induce apoptosis in some tumor cells by ery in the GAL4 system by 90-fold; a 50-fold increase was observed affecting alternate splicing of RNA (11). -

Intracrine Vascular Endothelial Growth Factor Signaling in Survival and Chemoresistance of Human Colorectal Cancer Cells
Oncogene (2011) 30, 1205–1212 & 2011 Macmillan Publishers Limited All rights reserved 0950-9232/11 www.nature.com/onc ORIGINAL ARTICLE Intracrine vascular endothelial growth factor signaling in survival and chemoresistance of human colorectal cancer cells S Samuel1, F Fan1,LHDang2, L Xia1, P Gaur3 and LM Ellis1,3 1Department of Cancer Biology, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA; 2Division of Hematology/Oncology, Department of Medicine, University of Florida, Gainesville, FL, USA and 3Department of Surgical Oncology, The University of Texas M. D. Anderson Cancer Center, Houston, TX, USA Although the effects of vascular endothelial growth factor proliferation of endothelial cells (reviewed in Brown (VEGF) on angiogenesis and vascular function are well et al., 1997). It has since been shown that VEGF can known, the effects of VEGF on tumor cell function also mediate a protective/survival effect on a number remain to be elucidated. We studied phenotypic changes in of cell types, including endothelial cells, embryonic human colorectal cancer (CRC) cells with homozygous stem cells and hematopoietic stem cells (Ferrara et al., deletion of VEGF alleles to determine the potential direct 1996; Gerber et al., 1998, 2002). In addition, recent role of VEGF on tumor cell function. Loss of VEGF studies have shown that VEGF acts as an autocrine/ expression led to significantly decreased cell growth and paracrine growth and survival factor for tumor cells that increased spontaneous apoptosis in CRC cells (Po0.01). express VEGF receptor (Bachelder et al., 2001; Pidgeon Loss of VEGF also increased the in vitro sensitivity of et al., 2001; Dias et al., 2002; Santos and Dias, 2004; cells to the cytotoxic effects of the chemotherapeutic drug Vincent et al., 2005; Barr et al., 2008; Calvani et al., 5-fluorouracil, as shown by increased apoptosis (Po0.05). -

Response to Androgens and Androgen Receptor Antagonists in the Presence of Cytokines in Prostate Cancer
cancers Review Response to Androgens and Androgen Receptor Antagonists in the Presence of Cytokines in Prostate Cancer Zoran Culig Experimental Urology, Department of Urology, Medical University of Innsbruck, Anichstrasse 35, A-6020 Innsbruck, Austria; [email protected] Simple Summary: Prostate cancer is the most frequently diagnosed non-cutaneous tumor in men in the Western world. Therapy for non-organ confined prostate cancer includes anti-androgens such as bicalutamide, enzalutamide and darolutamide. The androgen receptor is expressed during tumor initiation and progression. Androgen receptor could be activated by interleukins, which are produced by blood cells and adjacent stroma. These cytokines may affect response of tumor cells to anti-androgenic drugs, which are commonly used in prostate cancer therapy. There are several experimental studies showing an effect of anti-cytokine therapies in prostate cancer. However, the clinical translation is limited and more clinical trials are needed to improve action of anti-androgens in prostate cells which are stimulated by cytokines. Abstract: Non-steroidal anti-androgens have a major role in the treatment of non-localized prostate cancer. Interleukins are involved in the regulation of many cellular functions in prostate cancer and also modify cellular response to anti-androgens. A specific role of selected IL is presented in this review. IL-8 is a cytokine expressed in prostate cancer tissue and microenvironment and promotes proliferation and androgen receptor-mediated transcription. In contrast, IL-1 displays negative effects on expression of androgen receptor and its target genes. A subgroup of prostate cancers show Citation: Culig, Z. Response to neuroendocrine differentiation, which may be in part stimulated by androgen ablation. -

Role of Talin1 Phosphorylation in Beta1 Integrin Activation and Prostate Cancer Metastasis
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 5-2014 ROLE OF TALIN1 PHOSPHORYLATION IN BETA1 INTEGRIN ACTIVATION AND PROSTATE CANCER METASTASIS Jung-Kang Jin Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Cancer Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Jin, Jung-Kang, "ROLE OF TALIN1 PHOSPHORYLATION IN BETA1 INTEGRIN ACTIVATION AND PROSTATE CANCER METASTASIS" (2014). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 445. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/445 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. ROLE OF TALIN1 PHOSPHORYLATION IN β1 INTEGRIN ACTIVATION AND PROSTATE CANCER METASTASIS by Jung-Kang Jin, M.S. APPROVED: _____________________________________ -
Exploring the Intracrine Functions of VEGF-A
biomolecules Review Exploring the Intracrine Functions of VEGF-A Sophie Wiszniak and Quenten Schwarz * Centre for Cancer Biology, University of South Australia and SA Pathology, Adelaide, SA 5000, Australia; [email protected] * Correspondence: [email protected]; Tel.: +61-8-8302-7834 Abstract: Vascular endothelial growth factor A (VEGF-A or VEGF) is a highly conserved secreted signalling protein best known for its roles in vascular development and angiogenesis. Many non- endothelial roles for VEGF are now established, with the discovery that VEGF and its receptors VEGFR1 and VEGFR2 are expressed in many non-vascular cell-types, as well as various cancers. In addition to secreted VEGF binding to its receptors in the extracellular space at the cell membrane (i.e., in a paracrine or autocrine mode), intracellularly localised VEGF is emerging as an important signalling molecule regulating cell growth, survival, and metabolism. This intracellular mode of signalling has been termed “intracrine”, and refers to the direct action of a signalling molecule within the cell without being secreted. In this review, we describe examples of intracrine VEGF signalling in regulating cell growth, differentiation and survival, both in normal cell homeostasis and development, as well as in cancer. We further discuss emerging evidence for the molecular mechanisms underpinning VEGF intracrine function, as well as the implications this intracellular mode of VEGF signalling may have for use and design of anti-VEGF cancer therapeutics. Keywords: VEGF; VEGF-A; intracrine; autocrine; intracellular; signaling; cell survival; apoptosis; cancer; anti-VEGF therapy Citation: Wiszniak, S.; Schwarz, Q. Exploring the Intracrine Functions of 1. -

Intracrine Testosterone Activation in Human Pancreatic Β Cells Stimulates Insulin
Page 1 of 51 Diabetes Intracrine testosterone activation in human pancreatic β cells stimulates insulin secretion. Weiwei Xu1, Lina Schiffer2, M.M. Fahd Qadir1, Yanqing Zhang1, James Hawley3, Paula Mota De Sa1 Brian G Keevil3, Hongju Wu1, Wiebke Arlt2,4, Franck Mauvais-Jarvis1,5 1Department of Medicine, Section of Endocrinology and Metabolism, Tulane University Health Sciences Center, 2Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, B15 2TT, UK; 3Department of Clinical Biochemistry, Wythenshawe Hospital, Manchester National Health Service Foundation Trust, Manchester, M23 9LT, UK; 3National Institute for Health Research (NIHR) Birmingham Biomedical Research Centre, University of Birmingham and University Hospitals Birmingham NHS Foundation Trust, Birmingham, B15 3GW, UK; 2Southeast Louisiana Veterans Affairs Healthcare System, New Orleans, LA 70112, USA; Running title: Intracrine testosterone metabolism and insulin secretion Tweet: Human pancreatic #betacells locally produce active androgens and estrogens from circulating testosterone to stimulates insulin production #diabetes @FMauvaisJarvis @TulaneMedicine *Correspondance: Franck Mauvais-Jarvis, MD, Ph.D. Department of Medicine, Section of Endocrinology and Metabolism, Tulane University Health Sciences Center, 1430 Tulane Avenue, New Orleans, LA 70112, U.S.A. Email: [email protected] 1 Diabetes Publish Ahead of Print, published online August 27, 2020 Diabetes Page 2 of 51 Abstract Testosterone (T) affects β cell function in men and women. T is a pro-hormone that undergoes intracrine conversion in target tissues to the potent androgen dihydrotestosterone (DHT) via the enzyme 5α-reductase (5α-R), or to the active estrogen 17β-estradiol (E2) via the aromatase enzyme. Using male and female human pancreas sections, we show that the 5α-R type1 isoform (SRD5A1) and aromatase are expressed in male and female β cells.