Role of Talin1 Phosphorylation in Beta1 Integrin Activation and Prostate Cancer Metastasis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biological Effect of the Interaction Between Mental Retardation Related Protein
生物化学与生物物理进展 ProgressinBiochemistryandBiophysics 2013,40(11):1124~1131 www.pibb.ac.cn BiologicalEffectofTheInteractionBetween MentalRetardationRelatedProtein (FXR1P)andCMAS* MAYun1)**,QINLing-Xue1)**,DONGXiao1),LIBin-Yuan1),WANGChang-Bo1), XUCan2),WANGSan-Hu1),HEShu-Ya1)*** (1) DepartmentofBiochemistry&Biology,UniversityofSouthChina,Hengyang 421001,China; 2) DepartmentofCardiologyofTheFisrtAffiliatedHospitalofUniversityofSouthChina,Hengyang 421001,China) Abstract FragileXsyndrome(FXS)isageneticmentalretardationdisease,withincidencesecondonlytotrisomy21syndrome. FragileXmentalretardationprotein(FMRP),isthecausativefactorofFXSandencodedbytheFragileXmentalretardation1(FMR1) gene,whichiswidelyexpressedincellsofthenerve,muscle,andtestes.FragileXrelatedprotein1(FXR1P)isencodedbya homologousgeneto FMR1———FragileXrelatedgene1(FXR1)andcaninteractwithproteinsandRNAs.Manyillnesseswere involvedinthealteredexpressionof FXR1.TounderstandthebiologicaleffectoftheinteractionbetweenFXR1PandCMAS,we constructedaFXR1 overexpressionvectorandinvestigateditsexpressioninPC12(theratpheochromocytoma)cellsandVSMC (vascularsmoothmusclecell)andtheeffectoftheoverexpressiononcellmorphologyandseveralcellprocessesrelatedto CMP-N-acetylneuraminicacidsynthetase(CMAS)activity.Wedemonstratethattheoverexpressionof FXR1 genecanincreaseactivity ofCMASinPC12cellsandprovideacertaindegreegrowthprotectionforthatcells.Thus,itsuggestsFXR1Pisatissue-specific regulatortoaltertheconcentrationofGM1inPC12cells,butnotinVSMC. Keywords FXR1P,CMAS,GM1,biologicaleffect,PC12,VSMC DOI:10.3724/SP.J.1206.2013.00004 -

An Intracrine View of Sex Steroids, Immunity, and Metabolic Regulation
Review An intracrine view of sex steroids, immunity, and metabolic regulation Katya B. Rubinow ABSTRACT Background: Over the past two decades, parallel recognition has grown of the importance of both sex steroids and immune activity in metabolic regulation. More recently, these discrete areas have been integrated in studies examining the metabolic effects of sex steroid immunomodulation. Implicit in these studies has been a traditional, endocrine model of sex steroid delivery from the gonads to target cells, including immune cells. Thus, research to date has focused on the metabolic effects of sex steroid receptor signaling in immune cells. This endocrine model, however, overlooks the extensive capacity of immune cells to generate and metabolize sex steroids, enabling the production of sex steroids for intracrine signaling e that is, sex steroid production for signaling within the cell of origin. Intracrine function allows highly cell-autonomous regulation of sex steroid exposure, and sex steroid secretion by immune cells could confer paracrine signaling effects in neighboring cells within metabolic tissues. In this review, immune cell intracrinology will denote sex steroid production within immune cells for either intracrine or paracrine signaling. This intracrine capacity of immune cells has been well established, and prior work has supported its importance in autoimmune disorders, trauma, and cancer. The potential relevance of immune cell intracrine function to the regulation of energy balance, body weight, body composition, and insulin sensitivity has yet to be explored. Scope of review: The following review will detail findings to date regarding the steroidogenic and steroid metabolizing capacity of immune cells, the regulation of immune cell intracrine function, and the biological effects of immune-derived sex steroids, including the clinical relevance of immune cell intracrinology in fields other than metabolism. -

Sulfation Through the Looking Glass—Recent Advances in Sulfotransferase Research for the Curious
The Pharmacogenomics Journal (2002) 2, 297–308 & 2002 Nature Publishing Group All rights reserved 1470-269X/02 $25.00 www.nature.com/tpj REVIEW Sulfation through the looking glass—recent advances in sulfotransferase research for the curious MWH Coughtrie ABSTRACT Members of the cytosolic sulfotransferase (SULT) superfamily catalyse the Department of Molecular & Cellular Pathology, sulfation of a multitude of xenobiotics, hormones and neurotransmitters. University of Dundee, Ninewells Hospital & Humans have at least 10 functional SULT genes, and a number of recent Medical School, Dundee, Scotland, UK advances reviewed here have furthered our understanding of SULT function. Correspondence: Analysis of expression patterns has shown that sulfotransferases are highly MWH Coughtrie, Department of expressed in the fetus, and SULTs may in fact be a major detoxification Molecular & Cellular Pathology, University enzyme system in the developing human. The X-ray crystal structures of of Dundee, Ninewells Hospital and three SULTs have been solved and combined with mutagenesis experiments Medical School, Dundee DD1 9SY, Scotland, UK. and molecular modelling, they have provided the first clues as to the factors Tel: +44 (0)1382 632510 that govern the unique substrate specificities of some of these enzymes. In Fax: +44 (0)1382 640320 the future these and other studies will facilitate prediction of the fate of E-mail: [email protected] chemicals metabolised by sulfation. Variation in sulfation capacity may be important in determining an individual’s response to xenobiotics, and there has been an explosion in information on sulfotransferase polymorphisms and their functional consequences, including the influence of SULT1A1 genotype on susceptibility to colorectal and breast cancer. -
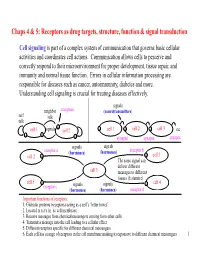
Chaps 4 & 5: Receptors As Drug Targets, Structure, Function & Signal Transduction
Chaps 4 & 5: Receptors as drug targets, structure, function & signal transduction Cell signaling is part of a complex system of communication that governs basic cellular activities and coordinates cell actions. Communication allows cells to perceive and correctly respond to their microenvironment for proper development, tissue repair, and immunity and normal tissue function. Errors in cellular information processing are responsible for diseases such as cancer, autoimmunity, diabetes and more. Understanding cell signaling is crucial for treating diseases effectively. signals neighbor receptors (neurotransmitters) self talk talk cell 1 signals cell 2 cell 1 cell 2 cell 3 etc. synapse synapse synapse signals signals receptor a (hormones) (hormones) receptor b cell 2 cell 3 The same signal can deliver different cell 1 messages to different tissues (histamine). cell 5 cell 4 receptor c signals signals (hormones) (hormones) receptor d Important functions of receptors: 1. Globular proteins (receptors) acting as a cell’s ‘letter boxes’ 2. Located mostly in the cell membrane 3. Receive messages from chemical messengers coming from other cells 4. Transmit a message into the cell leading to a cellular effect 5. Different receptors specific for different chemical messengers 6. Each cell has a range of receptors in the cell membrane making it responsive to different chemical messengers 1 © Oxford University Press, 2013 Signals can be divided into the 5 catagories below. Signal carriers (S) have to reach the proper receptors (R) for the message to be recieved. Intracrine signals are produced by the target cell that stay within the target cell. cell a S = signal S R R = receptor Autocrine signals are produced by the target cell, are secreted, and affect the target cell itself via receptors. -

Loss of Mouse Cardiomyocyte Talin-1 and Talin-2 Leads to Β-1 Integrin
Loss of mouse cardiomyocyte talin-1 and talin-2 leads PNAS PLUS to β-1 integrin reduction, costameric instability, and dilated cardiomyopathy Ana Maria Mansoa,b,1, Hideshi Okadaa,b, Francesca M. Sakamotoa, Emily Morenoa, Susan J. Monkleyc, Ruixia Lia, David R. Critchleyc, and Robert S. Rossa,b,1 aDivision of Cardiology, Department of Medicine, University of California at San Diego School of Medicine, La Jolla, CA 92093; bCardiology Section, Department of Medicine, Veterans Administration Healthcare, San Diego, CA 92161; and cDepartment of Molecular Cell Biology, University of Leicester, Leicester LE1 9HN, United Kingdom Edited by Kevin P. Campbell, Howard Hughes Medical Institute, University of Iowa, Iowa City, IA, and approved May 30, 2017 (received for review January 26, 2017) Continuous contraction–relaxation cycles of the heart require ognized as key mechanotransducers, converting mechanical per- strong and stable connections of cardiac myocytes (CMs) with turbations to biochemical signals (5, 6). the extracellular matrix (ECM) to preserve sarcolemmal integrity. The complex of proteins organized by integrins has been most CM attachment to the ECM is mediated by integrin complexes commonly termed focal adhesions (FA) by studies performed in localized at the muscle adhesion sites termed costameres. The cells such as fibroblasts in a 2D environment. It is recognized that ubiquitously expressed cytoskeletal protein talin (Tln) is a compo- this structure is important for organizing and regulating the me- nent of muscle costameres that links integrins ultimately to the chanical and signaling events that occur upon cellular adhesion to sarcomere. There are two talin genes, Tln1 and Tln2. Here, we ECM (7, 8). -

Novel Roles for Scleraxis in Regulating Adult Tenocyte Function Anne E
Nichols et al. BMC Cell Biology (2018) 19:14 https://doi.org/10.1186/s12860-018-0166-z RESEARCH ARTICLE Open Access Novel roles for scleraxis in regulating adult tenocyte function Anne E. C. Nichols1, Robert E. Settlage2, Stephen R. Werre3 and Linda A. Dahlgren1* Abstract Background: Tendinopathies are common and difficult to resolve due to the formation of scar tissue that reduces the mechanical integrity of the tissue, leading to frequent reinjury. Tenocytes respond to both excessive loading and unloading by producing pro-inflammatory mediators, suggesting that these cells are actively involved in the development of tendon degeneration. The transcription factor scleraxis (Scx) is required for the development of force-transmitting tendon during development and for mechanically stimulated tenogenesis of stem cells, but its function in adult tenocytes is less well-defined. The aim of this study was to further define the role of Scx in mediating the adult tenocyte mechanoresponse. Results: Equine tenocytes exposed to siRNA targeting Scx or a control siRNA were maintained under cyclic mechanical strain before being submitted for RNA-seq analysis. Focal adhesions and extracellular matrix-receptor interaction were among the top gene networks downregulated in Scx knockdown tenocytes. Correspondingly, tenocytes exposed to Scx siRNA were significantly softer, with longer vinculin-containing focal adhesions, and an impaired ability to migrate on soft surfaces. Other pathways affected by Scx knockdown included increased oxidative phosphorylation and diseases caused by endoplasmic reticular stress, pointing to a larger role for Scx in maintaining tenocyte homeostasis. Conclusions: Our study identifies several novel roles for Scx in adult tenocytes, which suggest that Scx facilitates mechanosensing by regulating the expression of several mechanosensitive focal adhesion proteins. -

Cell Plasticity and Prostate Cancer: the Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance
cancers Review Cell Plasticity and Prostate Cancer: The Role of Epithelial–Mesenchymal Transition in Tumor Progression, Invasion, Metastasis and Cancer Therapy Resistance Sofia Papanikolaou, Aikaterini Vourda, Spyros Syggelos and Kostis Gyftopoulos * Department of Anatomy, University of Patras School of Medicine, Rion, 26504 Patras, Greece; [email protected] (S.P.); [email protected] (A.V.); [email protected] (S.S.) * Correspondence: [email protected] Simple Summary: Although epithelial-to-mesenchymal transition (EMT) is a well-known cellular process involved during normal embryogenesis and wound healing, it also has a dark side; it is a complex process that provides tumor cells with a more aggressive phenotype, facilitating tumor metastasis and even resistance to therapy. This review focuses on the key pathways of EMT in the pathogenesis of prostate cancer and the development of metastases and evasion of currently available treatments. Abstract: Prostate cancer, the second most common malignancy in men, is characterized by high heterogeneity that poses several therapeutic challenges. Epithelial–mesenchymal transition (EMT) is a dynamic, reversible cellular process which is essential in normal embryonic morphogenesis and Citation: Papanikolaou, S.; Vourda, wound healing. However, the cellular changes that are induced by EMT suggest that it may also A.; Syggelos, S.; Gyftopoulos, K. Cell play a central role in tumor progression, invasion, metastasis, and resistance to current therapeutic Plasticity and Prostate Cancer: The options. These changes include enhanced motility and loss of cell–cell adhesion that form a more Role of Epithelial–Mesenchymal aggressive cellular phenotype. Moreover, the reverse process (MET) is a necessary element of the Transition in Tumor Progression, Invasion, Metastasis and Cancer metastatic tumor process. -

Molecular Interactions of Talin and Actin: a Study on the Specific Domains of Talin Involved in the Interactions Ho-Sup Lee Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2002 Molecular interactions of talin and actin: a study on the specific domains of talin involved in the interactions Ho-Sup Lee Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Biochemistry Commons, Cell Biology Commons, and the Molecular Biology Commons Recommended Citation Lee, Ho-Sup, "Molecular interactions of talin and actin: a study on the specific domains of talin involved in the interactions " (2002). Retrospective Theses and Dissertations. 528. https://lib.dr.iastate.edu/rtd/528 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

Comparative Proteomic Analysis Identifies Epha2 As a Specific Cell
International Journal of Molecular Sciences Article Comparative Proteomic Analysis Identifies EphA2 as a Specific Cell Surface Marker for Wharton’s Jelly-Derived Mesenchymal Stem Cells Ashraf Al Madhoun 1,2,* , Sulaiman K. Marafie 3 , Dania Haddad 2, Motasem Melhem 2, Mohamed Abu-Farha 3 , Hamad Ali 2,4 , Sardar Sindhu 1 , Maher Atari 5 and Fahd Al-Mulla 2 1 Department of Animal and Imaging Core Facilities, Dasman Diabetes Institute, Dasman 15462, Kuwait; [email protected] 2 Department of Genetics and Bioinformatics, Dasman Diabetes Institute, Dasman 15462, Kuwait; [email protected] (D.H.); [email protected] (M.M.); [email protected] (H.A.); [email protected] (F.A.-M.) 3 Department of Biochemistry and Molecular Biology, Dasman Diabetes Institute, Dasman 15462, Kuwait; sulaiman.marafi[email protected] (S.K.M.); [email protected] (M.A.-F.) 4 Department of Medical Laboratory Sciences, Faculty of Allied Health Sciences, Health Sciences Center (HSC), Kuwait University, Jabriya 046302, Kuwait 5 Medical-Surgical Pathology Department, Regenerative Medicine Research Institute, Universitat Internacional de Catalunya, 08195 Barcelona, Spain; [email protected] * Correspondence: [email protected] Received: 28 June 2020; Accepted: 1 September 2020; Published: 3 September 2020 Abstract: Wharton’s jelly-derived mesenchymal stem cells (WJ-MSCs) are a valuable tool in stem cell research due to their high proliferation rate, multi-lineage differentiation potential, and immunotolerance properties. However, fibroblast impurity during WJ-MSCs isolation is unavoidable because of morphological similarities and shared surface markers. Here, a proteomic approach was employed to identify specific proteins differentially expressed by WJ-MSCs in comparison to those by neonatal foreskin and adult skin fibroblasts (NFFs and ASFs, respectively). -

Reticular Adhesions: a New Class of Adhesion Complex That Mediates Cell-Matrix Attachment During Mitosis
bioRxiv preprint doi: https://doi.org/10.1101/234237; this version posted December 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Reticular adhesions: A new class of adhesion complex that mediates cell-matrix attachment during mitosis John G. Lock1,2,*, Matthew C. Jones3,#, Janet A. Askari3,#, Xiaowei Gong2,#, Anna Oddone4,5, Helene Olofsson2, Sara Göransson2, Melike Lakadamyali5,6, Martin J. Humphries3, Staffan Strömblad2,* 1School of Medical Sciences, University of New South Wales, Sydney 2052, Australia 2Department of Biosciences and Nutrition, Karolinska Institutet, S-141 83 Huddinge, Sweden 3Wellcome Trust Centre for Cell-Matrix Research, Faculty of Biology, Medicine and Health, University of Manchester, Manchester M13 9PT, UK 4Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain 5ICFO, Institut de Ciencies Fotoniques, Mediterranean Technology Park, The Barcelona Institute of Science and Technology, 08860 Castelldefels, Barcelona, Spain 6Perelman School of Medicine, Department of Physiology, University of Pennsylvania, Philadelphia PA 19104, USA # These authors contributed equally to this study * Correspondence: [email protected]; [email protected] Staffan Strömblad John Lock Karolinska Institutet University of New South Wales, Novum, Hälsovägen 7-9, School of Medical Sciences, SE-141 83 Huddinge Kensington 2052 Sydney, Sweden Australia email: [email protected] email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/234237; this version posted December 14, 2017. -

Fibroblasts from the Human Skin Dermo-Hypodermal Junction Are
cells Article Fibroblasts from the Human Skin Dermo-Hypodermal Junction are Distinct from Dermal Papillary and Reticular Fibroblasts and from Mesenchymal Stem Cells and Exhibit a Specific Molecular Profile Related to Extracellular Matrix Organization and Modeling Valérie Haydont 1,*, Véronique Neiveyans 1, Philippe Perez 1, Élodie Busson 2, 2 1, 3,4,5,6, , Jean-Jacques Lataillade , Daniel Asselineau y and Nicolas O. Fortunel y * 1 Advanced Research, L’Oréal Research and Innovation, 93600 Aulnay-sous-Bois, France; [email protected] (V.N.); [email protected] (P.P.); [email protected] (D.A.) 2 Department of Medical and Surgical Assistance to the Armed Forces, French Forces Biomedical Research Institute (IRBA), 91223 CEDEX Brétigny sur Orge, France; [email protected] (É.B.); [email protected] (J.-J.L.) 3 Laboratoire de Génomique et Radiobiologie de la Kératinopoïèse, Institut de Biologie François Jacob, CEA/DRF/IRCM, 91000 Evry, France 4 INSERM U967, 92260 Fontenay-aux-Roses, France 5 Université Paris-Diderot, 75013 Paris 7, France 6 Université Paris-Saclay, 78140 Paris 11, France * Correspondence: [email protected] (V.H.); [email protected] (N.O.F.); Tel.: +33-1-48-68-96-00 (V.H.); +33-1-60-87-34-92 or +33-1-60-87-34-98 (N.O.F.) These authors contributed equally to the work. y Received: 15 December 2019; Accepted: 24 January 2020; Published: 5 February 2020 Abstract: Human skin dermis contains fibroblast subpopulations in which characterization is crucial due to their roles in extracellular matrix (ECM) biology. -

Environmental Sensing Through Focal Adhesions
FOCUS ON MECHANOTRANSDUCTREVIEWSION Environmental sensing through focal adhesions Benjamin Geiger*, Joachim P. Spatz‡ and Alexander D. Bershadsky* Abstract | Recent progress in the design and application of artificial cellular microenvironments and nanoenvironments has revealed the extraordinary ability of cells to adjust their cytoskeletal organization, and hence their shape and motility, to minute changes in their immediate surroundings. Integrin-based adhesion complexes, which are tightly associated with the actin cytoskeleton, comprise the cellular machinery that recognizes not only the biochemical diversity of the extracellular neighbourhood, but also its physical and topographical characteristics, such as pliability, dimensionality and ligand spacing. Here, we discuss the mechanisms of such environmental sensing, based on the finely tuned crosstalk between the assembly of one type of integrin-based adhesion complex, namely focal adhesions, and the forces that are at work in the associated cytoskeletal network owing to actin polymerization and actomyosin contraction. FIG. 1 Extracellular matrix Environmental sensing by living cells displays many growth, differentiation or programmed cell death). (ECM). The complex, features that are usually attributed to ‘intelligent systems’. shows the capacity of cells to respond to variations in multimolecular material that A cell can sense and respond to a wide range of ext ernal multiple surface parameters, including ECM specifi- surrounds cells. The ECM signals, both chemical and physical, it can integrate city, adhesive ligand density, surface compliance and comprises a scaffold on which tissues are organized, provides and analyse this information and, as a consequence, it dimensionality, by altering cell shape and cytoskeletal cellular microenvironments can change its morphology, dynamics, behaviour and, organization, and by modulating the adhesion sites.