UNIVERSITY of CALIFORNIA RIVERSIDE Electrochemical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

221 Gaas GRADED A
USOO6403874B1 (12) United States Patent (10) Patent No.: US 6,403,874 B1 Shakouri et al. (45) Date of Patent: *Jun. 11, 2002 (54) HIGH-EFFICIENCY HETEROSTRUCTURE OTHER PUBLICATIONS THERMONIC COOLERS N N. W. Ashcroft, et al., Solid State Physics, manual, 1976, pp. (75) Inventors: Ali Shakouri, Santa Cruz; John E. 318-319, 320-321, 362-363. Bowers, Santa Barbara, both of CA D. A. Broido et al., “Effect of Superlattice structure on the (US) th ermoelectriclectric fiIlgure OIf meril:t: , Theline Am erican PhPinySIca 1 Society (Physical Review B.), vol. 51, No. 19, May 15, (73) Assignee: The Regents of the University of 1995, pp. 13797-800. California, Oakland, CA (US) D. A. Broido et al., “Comment of Use of quantum well Superlattices to obtain high figure of merit from nonconven (*) Notice: Subject to any disclaimer, the term of this tional thermoelectric materials”, Appl. Phys. Lett. 63, 3230 patent is extended or adjusted under 35 (1993), Applied Physics Letters, vol. 67, No. 8, Aug. 21, U.S.C. 154(b) by 0 days. 1995, pp. 1170–1171. D. A. Broido et al., “Thermoelectric figure or merit of This patent is Subject to a terminal dis- quantum wire Superlattices”, Applied Physics Letters, Jul. 3, claimer. 1995, vol. 67, No. 1, 100-102. (21) Appl. No.: 09/441,787 (List continued on next page.) (22) Filed: Nov. 17, 1999 Primary Examiner—Bruce F. Bell ASSistant Examiner Thomas H Parsons Related U.S. Application Data (74) Attorney, Agent, or Firm-Gates & Cooper LLP (60) Pisional application No. 60/109,342, filed on Nov. -

Preparation of Bismuth Telluride Specimens for TEM
Preparation of Bismuth Telluride Specimens for TEM Mark Homer1, Douglas L. Medlin2 1,2. Sandia National Laboratories, Energy Nanomaterials Dept., Livermore CA, USA Bismuth telluride (Bi2Te3) and its alloys are an important class of thermoelectric material. How well a thermoelectric material works is dependent on a variety of factors such as electrical and thermal conductivity and the Seeback coefficient. Because the electrical and thermal conductivity can be affected by defects in the material, there is much interest in the basic understanding the microstructures of these materials [1]. There are challenges in preparation of TEM specimens from telluride-based materials due to their sensitivity to ion-milling artifacts. For instance, nanoscale defect arrangements have been shown to form in lead telluride (PbTe) specimens prepared under aggressive ion milling conditions if cooling and power density is not suitably controlled [2]. In this presentation we discuss methods and conditions for preparing TEM specimens of Bi2Te3 specimens considering both ion-milling and electropolishing techniques. In all cases TEM specimens were mechanically pre-thinned using conventional mechanical dimpling and polishing techniques prior to final thinning to electron transparency. The ion-milled specimens were prepared with Ar+ ion sputtering using a Fischione Model 1010 ion mill with LN cooling. The electropolished specimens were prepared using a Fischione Model 120 electropolisher and an electrolyte consisting of 53% water, 38% glycerol, 5% sodium hydroxide, and 4% tartaric acid. The electrolyte was set in an ice bath and cooled to 2° C, and electropolished at 25V and 35mA. Figures 1 and 2 show dark-field TEM micrographs comparing ion milled and electropolished Bi2Te3 specimens. -

Alfa Laval Black and Grey List, Rev 14.Pdf 2021-02-17 1678 Kb
Alfa Laval Group Black and Grey List M-0710-075E (Revision 14) Black and Grey list – Chemical substances which are subject to restrictions First edition date. 2007-10-29 Revision date 2021-02-10 1. Introduction The Alfa Laval Black and Grey List is divided into three different categories: Banned, Restricted and Substances of Concern. It provides information about restrictions on the use of Chemical substances in Alfa Laval Group’s production processes, materials and parts of our products as well as packaging. Unless stated otherwise, the restrictions on a substance in this list affect the use of the substance in pure form, mixtures and purchased articles. - Banned substances are substances which are prohibited1. - Restricted substances are prohibited in certain applications relevant to the Alfa Laval group. A restricted substance may be used if the application is unmistakably outside the scope of the legislation in question. - Substances of Concern are substances of which the use shall be monitored. This includes substances currently being evaluated for regulations applicable to the Banned or Restricted categories, or substances with legal demands for monitoring. Product owners shall be aware of the risks associated with the continued use of a Substance of Concern. 2. Legislation in the Black and Grey List Alfa Laval Group’s Black and Grey list is based on EU legislations and global agreements. The black and grey list does not correspond to national laws. For more information about chemical regulation please visit: • REACH Candidate list, Substances of Very High Concern (SVHC) • REACH Authorisation list, SVHCs subject to authorization • Protocol on persistent organic pollutants (POPs) o Aarhus protocol o Stockholm convention • Euratom • IMO adopted 2015 GUIDELINES FOR THE DEVELOPMENT OF THE INVENTORY OF HAZARDOUS MATERIALS” (MEPC 269 (68)) • The Hong Kong Convention • Conflict minerals: Dodd-Frank Act 1 Prohibited to use, or put on the market, regardless of application. -

Electronic Properties of Lead Telluride Quantum Wells
Electronic Properties of Lead Telluride Quantum Wells Liza Mulder Smith College 2013 NSF/REU Program Physics Department, University of Notre Dame Advisors: Profs. Jacek Furdyna, Malgorzata Dobrowolska, and Xinyu Liu Thanks also to: Rich Pimpinella, Joseph Hagmann, and Xiang Li Abstract My research this summer has focused on measuring the electronic properties of quantum wells that combine two families of semiconductors, specifically Cadmium Telluride and Lead Telluride (CdTe/PbTe/CdTe), whose interfaces hold promise of Topological Crystalline Insulator (TCI) surface states. The samples used in my study are grown by Molecular Beam Epitaxy, and measured using Magneto Transport experiments. I have found that samples grown on higher temperature substrates have higher charge carrier mobilities and lower carrier concentrations, while the resistivity shows no significant change. These results will be used as a guide toward designing future structures for observing and characterizing TCI surface states. Introduction Semiconductors have many applications in electronic devices, and as these devices become smaller and smaller, so do their components. Hence the importance of understanding the complex surface states of semiconductors. Some of these surfaces are also possible examples of Topological Insulators (TIs) or Topological Crystalline Insulators (TCIs). By studying semiconductors in the form of thin films, quantum wells, nanowires, and quantum dots, researchers hope to better understand semiconductors in this form, to measure the electronic and magnetic properties of their surface states, and to discover new examples of TIs and TCIs. This summer I studied the electronic properties of Cadmium Telluride - Lead Telluride (CdTe/PbTe:Bi/CdTe) quantum well structures. The samples are grown by Molecular Beam Epitaxy, and measured using Magneto Transport. -

Development of Si-Based High Efficiency Thermoelectric Materials
Impact Objectives • Investigate potential non-toxic, abundant bases with thermoelectric properties • Develop high-efficiency silicon-based thermoelectric materials Generating electricity from waste heat Associate Professor Ken Kurosaki describes the potential of thermoelectric materials to improve energy efficiency by turning waste heat into an inexpensive source of electrical power nuclear materials and TE material converts semiconductor materials such as silicon, it to usable electricity. The performance is to alter the microstructure at the of a TE material is determined by the nanoscale level. This helps scatter and material’s figure of merit called zT, where block heat phonons, thereby lowering a higher zT correlates to a higher-efficiency thermal conductivity. We are also focusing TE module built from that material. Much on improving the Seebeck coefficient, research effort has been concentrated which is a measure of how much voltage is Professor Ken Kurosaki Sora-at Tanusilp on trying to achieve higher efficiency, but produced by the material in response to the only small improvements are being made. temperature difference across the material. Can you tell us a bit about high efficiency Our goal is to try new strategies involving thermoelectric materials? nanostructuring combined with mixed What is your background and how did valence states, to improve the performance you become involved in thermoelectric Thermoelectric materials can convert heat of TE materials and thus the efficiency of TE materials? to electricity and vice versa and thus can be modules. utilised as power generation or solid-state I started out in nuclear engineering, cooling modules. They have many potential Why are you concentrating on silicon particularly in the characterisation of uses and may greatly enhance fuel and compounds? nuclear fuels. -

Lead and Bismuth Chalcogenide Systems Hurr.Lnc Lru, Luxn L. Y. CH
American Mineralogist, Volume 79, pages 1159-l 166, 1994 Lead and bismuth chalcogenide systems Hurr.lNc Lru, Luxn L. Y. CH,c.Nc Department of Geology, University of Maryland, College Park, Maryland2O742,U-5.4. Ansrnlcr Phaserelations in the systemsof lead and bismuth chalcogenideswere examined in the temperature range of 500-900 "C. In the system PbS-PbSe-BirSr-BirSer,a complete series of solid solutions exists between PbnBioS,,(heyrovskyite) and PbnBioSe,r,whereas along the join from Pb.BiuS,,(lillanite) to PbrBiuSe,,there are two terminal solid solutions. The join PbBirSo-PbBirSeois characterizedby an extensivesolid solution basedon weibullite. Six extensive ranges of solid solution exist in the system PbSe-PbTe-BirSer-BirTer,and their compositionsfall along the joins in Pb(Se,Te):Bir(Se,Te),ratios of l:0, 2.7:1, 2:I, l:1, l:2, and0: I . New phasesand solid solutions in the systemsof lead and bismuth sulfide and telluride and selenide and telluride are all structurally related, and their structures are based on stacked layers of PbTe (two layers) and BirTe, (five layers). The total number of layers (N) in the unit cell of a phase, mPbTe'nBirTer, is expressedby the equation N : Z(2m * 5n), where Z is the number of formula units in the unit cell and can only be derived from experimental data. For the four phasesalong the galena-tetradymitejoin in the PbS- PbTe-BirSr-BirTe, system, PbrBirSrTer, PbrBioSrTeo,PbBirSrTer, -and PbBinS'Teohave N, Z, a, and c-respectively:9, l, q.2l x, and ft.j1 A;32,2,4.23 A, and 60.00A;21,3, 4.8 A. -

Synthesis and Characterization of Pbte Quantum Dots
City University of New York (CUNY) CUNY Academic Works Dissertations and Theses City College of New York 2011 Synthesis and characterization of PbTe quantum dots Faiza Anwar CUNY City College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/cc_etds_theses/61 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] Synthesis and Characterization of PbTe Quantum Dots A Thesis Presented to The Faculty of the Chemistry Program The City College of New York In (Partial) Fulfillment of the Requirements for the Degree Master of Arts by FAIZA ANWAR April 2011 Table of Contents 1. Introduction ......................................................................................................................... 3 1.2 Metal Chalcogenides and Synthesis ................................................................................ 6 1.3 Physical -Chemical Synthesis: .......................................................................................... 8 1.4 Lead Telluride (PbTe)- Physical and Chemical properties. .............................................. 13 1.5 Experimental Summary- Chemicals ............................................................................... 14 1.6 Experimental Set Up: ................................................................................................... 15 2.Instrumental Analysis: ....................................................................................................... -

Angstrom Sciences PVD Materials
40 S. Linden Street, Duquesne, PA 15110 USA Tel: (412) 469-8466 Fax: (412) 469-8511 www.angstromsciences.com PVD Materials Target Sputtering Targets, Pellets, Bonding Services Slugs, Starter Charges, Crucibles Recycling of precious metal All Configurations of Clips, scrap Rod, Tube, pieces and Wire Pure Elements / Noble Metals Aluminum, (Al) Gadolinium, (Gd) Nickel, (Ni) Tantalum, (Ta) Antimony, (Sb) Gallium, (Ga) Niobium, (Nb) Tellurium, (Te) Beryllium, (Be) Germanium, (Ge) Osmium, (Os) Terbium, (Tb) Bismuth, (Bi) Gold, (Au) Palladium, (Pd) Tin, (Sn) Boron, (B) Hafnium, (Hf) Platinum, (Pt) Titanium, (Ti) Cadmium, (Cd) Indium, (In) Praseodymium, (Pr) Tungsten, (W) Calcium, (Ca) Iridium, (Ir) Rhenium, (Re) Vanadium, (V) Carbon, (C) Iron, (Fe) Rhodium, (Rh) Ytterbium, (Yb) Cerium, (Ce) Lanthanum, (La) Ruthenium, (Ru) Yttrium, (Y) Chromium, (Cr) Lead, (Pb) Samarium, (Sm) Zinc, (Zn) Cobalt, (Co) Magnesium, (Mg) Selenium, (Se) Zirconium, (Zr) Copper, (Cu) Manganese, (Mn) Silicon, (Si) Erbium, (Er) Molybdenum, (Mo) Silver, (Ag) Precious Metal Alloys Gold Antimony, (Au/Sb) Gold Zinc, (Au/Zn) Gold Boron, (Au/B) Palladium Manganese, (Pd/Mn) Gold Copper, (Au/Cu) Palladium Nickel, (Pd/Ni) Gold Germanium, (Au/Ge) Platinum Palladium, (Pt/Pd) Gold Nickel, (Au/Ni) Palladium Rhenium, (Pd/Re) Gold Nickel Indium, (Au/Ni/In) Platinum Rhodium, (Pt/Rh) Gold Palladium, (Au/Pd) Silver Copper, (Ag/Cu) Gold Silicon, (Au/Si) Silver Gallium, (Ag/Ga) Gold Silver Platinum, (Au/Ag/Pt) Silver Gold, (Ag/Au) Gold Tantalum, (Au/Ta) Silver Palladium, (Ag/Pd) Gold Tin, -

Free Download
ALB Materials Inc 2360 Corporate Circle . Suite 400 Website: www.albmaterials.com Henderson, NV 89074-7739 E-mail: [email protected] Item No. Product Name CAS Number Formula Purity ALB‐semi‐AlSb Aluminum Antimonide [25152‐52‐7] AlSb 5N ALB‐semi‐Al2Se3 Aluminum Selenide [1302‐82‐5] Al2Se3 5N ALB‐semi‐Al2S3 Aluminum Sulfide [1302‐81‐4] Al2S3 5N ALB‐semi‐Al2Te3 Aluminum Telluride [12043‐29‐7] Al2Te3 5N ALB‐semi‐Sb Antimony [7440‐36‐0] Sb 5N, 6N ALB‐semi‐Sb2Se3 Antimony Selenide [1315‐05‐5] Sb2Se3 5N ALB‐semi‐Sb2S3 Antimony Sulfide [1345‐04‐6] Sb2S3 5N ALB‐semi‐Sb2Te3 Antimony Telluride [1327‐50‐0] Sb2Te3 4N, 5N ALB‐semi‐SbI3 Antimony(III) Iodide [7790‐44‐5] SbI3 4N ALB‐semi‐Sb2O3 Antimony(III) Oxide [1309‐64‐4] Sb2O3 5N ALB‐semi‐Sb2O5 Antimony(V) Oxide [1314‐60‐9] Sb2O5 5N ALB‐semi‐As2Se3 Arsenic Selenide [1303‐36‐2] As2Se3 5N ALB‐semi‐As2S3 Arsenic Sulfide [1303‐33‐9] As2S3 5N ALB‐semi‐Bi2O3 Bismuth Oxide [1304‐76‐3] Bi2O3 5N ALB‐semi‐Bi2Se3 Bismuth Selenide [12068‐69‐8] Bi2Se3 5N ALB‐semi‐Bi2S3 Bismuth Sulfide [1345‐07‐9] Bi2S3 5N ALB‐semi‐Bi2Te3 Bismuth Telluride [1304‐82‐1] Bi2Te3 4N, 5N ALB‐semi‐Cd Cadmium [7440‐43‐9]Inc Cd 5N, 6N, 7N ALB‐semi‐CdSb Cadmium Antimonide [12050‐27‐0] CdSb 5N ALB‐semi‐Cd3As2 Cadmium Arsenide [12006‐15‐4] Cd3As2 5N ALB‐semi‐CdSe Cadmium Selenide [1306‐24‐7] CdSe 4N, 5N ALB‐semi‐Cd2SnO4 Cadmium Stannate [12185‐56‐7] Cd2SnO4 5N ALB‐semi‐CdS Cadmium Sulfide [1306‐23‐6] CdS 4N, 5N ALB‐semi‐CdTe Cadmium Telluride [1306‐25‐8] CdTe 5N ALB‐semi‐Cu2S Copper Sulfide [22205‐45‐4] Cu2S 5N ALB‐semi‐GaSb Gallium Antimonide -

TOXICS RELEASE INVENTORY Guidance for Reporting the Lead and Lead Compounds Category
United States Environmental Office of Pollution Prevention and Toxics EPA 745-B-19-020 Protection Agency Washington, DC 20460 Revised February 2019 TOXICS RELEASE INVENTORY Guidance for Reporting the Lead and Lead Compounds Category Section 313 of the Emergency Planning and Community Right-to-Know Act of 1986 (EPCRA) requires certain facilities manufacturing, processing, or otherwise using listed toxic chemicals to report the annual quantity of such chemicals entering each environmental medium. Such facilities must also report pollution prevention and recycling data for such chemicals, pursuant to section 6607 of the Pollution Prevention Act, 42 U.S.C. 13106. EPCRA section 313 is also known as the Toxics Release Inventory (TRI). CONTENTS INTRODUCTION .............................................................................................................. 1 Section 1.1 What Are the Reporting (Activity) Thresholds? ................................................................ 1 Section 1.2 What Other Changes to the EPCRA Section 313 Reporting Requirements Apply to Lead and the Lead Compounds Category? ................................................................................................ 3 GUIDANCE FOR DETERMINING WHETHER REPORTING THRESHOLDS ARE EXCEEDED AND REPORTING ENVIRONMENTAL RELEASES OF LEAD AND LEAD COMPOUNDS ........................................................................................................................ 5 Section 2.1 General Guidance .............................................................................................................. -

Hot Wall Growth and Properties of Lead Telluride Films Doped by Germanium and Gallium
Semiconductor Physics, Quantum Electronics & Optoelectronics. 2000. V. 3, N 3. P. 295-299. PACS: 71.28, 71.55, 72.20.P, 72.20.M, 73.61.W Hot wall growth and properties of lead telluride films doped by germanium and gallium G.V. Lashkarev1*, M.V. Radchenko1, E.I. Slynko1, V.N. Vodopiyanov1, V.V. Asotsky1, V.M. Kaminsky1, G.V. Beketov2, E.V. Rengevich2 1 Institute for Problems of Material Science, Ukrainian National Academy of Sciences, Krzhizhanovskogo str.3, 03142 Kyiv, Ukraine Phone: (044) 444-3228; Fax: (044) 444-2131; e-mail: [email protected] 2 Institute for Semiconductor Physics, Ukrainian National Academy of Sciences, pr. Nauki 45, 252650 Kyiv, Ukraine Phone : (044) 265-5749; Fax: (044) 265-8344; e-mail: [email protected] Abstract. The researches results of films (PbTe)1-y(GaTe)y and (Pb1-xGexTe)1-y(GaTe)y of n-type conductivity with carrier concentration within (2⋅1017÷9⋅1018) cm-3 are represented. The films were growth by modified hot wall technology on barium fluoride substrate. A state of films surface was investigated by Atomic Force Microscope using both contact and taping modes. Hall coefficient, specific resistivity and thermoelectromotive force of films were measured in the temperature range 77-300 K. The investigation showed that the presence of germanium leads to an appearance of significant features on temperature dependencies of termoelectro- motive force which are connected with phase transition of displacement type. Doping gallium leads to n-type conductivity of films which increases with impurity concentration. The ioni- zation energies of Ga impurities levels (n - exp(-E/2kT)) were within 3÷10 meV. -
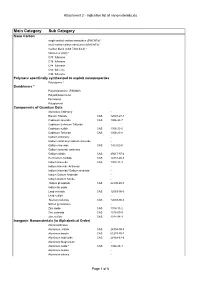
Attachment 2 - Indicative List of Nanomaterials.Xls
Attachment 2 - Indicative list of nanomaterials.xls Main Category Sub Category Nano Carbon single-walled carbon nanotubes (SWCNTs) * multi-walled carbon nanotubes (MWCNTs) * Carbon black (CAS 7440-44-0) * fullerenes (C60) * C70 fullerene C76 fullerene C84 fullerene C90 fullerene C94 fullerene Polymers specifically synthesized to exploit nanoproperties Polystyrene * Dendrimers * Polyamidoamine (PAMAM) Polyethylene imine Ferrocenyl Polyglycerol Components of Quantum Dots Aluminum Antinomy - Barium Titanate CAS 12047-27-7 Cadmium selenide CAS 1306-24-7 Cadmium Selenium Telluride Cadmium sulfide CAS 1306-23-6 Cadmium Telluride CAS 1306-25-8 Gallium Antimony - Gallium antimony/ Gallium arsenide - Gallium Arsenide CAS 1303-00-0 Gallium arsenide antimony Gallium nitride CAS 25617-97-4 Germanium carbide CAS 12334-26-8 Indium Arsenide CAS 1303-11-3 Indium Arsenide Antimony - Indium Arsenide/ Gallium arsenide - Indium Gallium Arsenide - Indium Gallium Nitride - Indium phosphide CAS 22398-80-7 Indium tin oxide - Lead selenide CAS 12069-00-0 Lead sulfide - Mercury telluride CAS 12068-90-5 Silicon germanium - Zinc oxide CAS 1314-13-2 Zinc selenide CAS 1315-09-9 Zinc sulfide CAS 1314-98-3 Inorganic Nanomaterials (in Alphabetical Order) Aluminosilicates - Aluminum nitride CAS 24304-00-5 Aluminum borate CAS 61279-70-7 Aluminum hydroxide CAS 20768-67-6 Aluminum Magnesium - Aluminum oxide * CAS 1344-28-1 Aluminum oxides - Aluminum silicate - Page 1 of 6 Attachment 2 - Indicative list of nanomaterials.xls Main Category Sub Category Aluminum Titanium Oxide