Analysis of the Detection of Organophosphate Pesticides in Aqueous Solutions Using Polymer-Coated Sh-Saw Devices
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
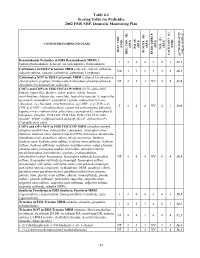
2002 NRP Section 6, Tables 6.1 Through
Table 6.1 Scoring Table for Pesticides 2002 FSIS NRP, Domestic Monitoring Plan } +1 0.05] COMPOUND/COMPOUND CLASS * ) (EPA) (EPA) (EPA) (EPA) (EPA) (FSIS) (FSIS) PSI (P) TOX.(T) L-1 HIST. VIOL. BIOCON. (B) {[( (2*R+P+B)/4]*T} REG. CON. (R) * ENDO. DISRUP. LACK INFO. (L) LACK INFO. {[ Benzimidazole Pesticides in FSIS Benzimidazole MRM (5- 131434312.1 hydroxythiabendazole, benomyl (as carbendazim), thiabendazole) Carbamates in FSIS Carbamate MRM (aldicarb, aldicarb sulfoxide, NA44234416.1 aldicarb sulfone, carbaryl, carbofuran, carbofuran 3-hydroxy) Carbamates NOT in FSIS Carbamate MRM (carbaryl 5,6-dihydroxy, chlorpropham, propham, thiobencarb, 4-chlorobenzylmethylsulfone,4- NT 4 1 3 NV 4 4 13.8 chlorobenzylmethylsulfone sulfoxide) CHC's and COP's in FSIS CHC/COP MRM (HCB, alpha-BHC, lindane, heptachlor, dieldrin, aldrin, endrin, ronnel, linuron, oxychlordane, chlorpyrifos, nonachlor, heptachlor epoxide A, heptachlor epoxide B, endosulfan I, endosulfan I sulfate, endosulfan II, trans- chlordane, cis-chlordane, chlorfenvinphos, p,p'-DDE, p, p'-TDE, o,p'- 3444NV4116.0 DDT, p,p'-DDT, carbophenothion, captan, tetrachlorvinphos [stirofos], kepone, mirex, methoxychlor, phosalone, coumaphos-O, coumaphos-S, toxaphene, famphur, PCB 1242, PCB 1248, PCB 1254, PCB 1260, dicofol*, PBBs*, polybrominated diphenyl ethers*, deltamethrin*) (*identification only) COP's and OP's NOT in FSIS CHC/COP MRM (azinphos-methyl, azinphos-methyl oxon, chlorpyrifos, coumaphos, coumaphos oxon, diazinon, diazinon oxon, diazinon met G-27550, dichlorvos, dimethoate, dimethoate -

Lifetime Organophosphorous Insecticide Use Among Private Pesticide Applicators in the Agricultural Health Study
Journal of Exposure Science and Environmental Epidemiology (2012) 22, 584 -- 592 & 2012 Nature America, Inc. All rights reserved 1559-0631/12 www.nature.com/jes ORIGINAL ARTICLE Lifetime organophosphorous insecticide use among private pesticide applicators in the Agricultural Health Study Jane A. Hoppin1, Stuart Long2, David M. Umbach3, Jay H. Lubin4, Sarah E. Starks5, Fred Gerr5, Kent Thomas6, Cynthia J. Hines7, Scott Weichenthal8, Freya Kamel1, Stella Koutros9, Michael Alavanja9, Laura E. Beane Freeman9 and Dale P. Sandler1 Organophosphorous insecticides (OPs) are the most commonly used insecticides in US agriculture, but little information is available regarding specific OP use by individual farmers. We describe OP use for licensed private pesticide applicators from Iowa and North Carolina in the Agricultural Health Study (AHS) using lifetime pesticide use data from 701 randomly selected male participants collected at three time periods. Of 27 OPs studied, 20 were used by 41%. Overall, 95% had ever applied at least one OP. The median number of different OPs used was 4 (maximum ¼ 13). Malathion was the most commonly used OP (74%) followed by chlorpyrifos (54%). OP use declined over time. At the first interview (1993--1997), 68% of participants had applied OPs in the past year; by the last interview (2005--2007), only 42% had. Similarly, median annual application days of OPs declined from 13.5 to 6 days. Although OP use was common, the specific OPs used varied by state, time period, and individual. Much of the variability in OP use was associated with the choice of OP, rather than the frequency or duration of application. -

NIOSH Method 5600: Organophosphorus Pesticides
ORGANOPHOSPHORUS PESTICIDES 5600 Formula: Table 1 MW: Table 1 CAS: Table 1 RTECS: Table 1 METHOD: 5600, Issue 1 EVALUATION: FULL Issue 1: 15 August 1994 OSHA : Table 2 PROPERTIES: Table 3 NIOSH: Table 2 ACGIH: Table 2 SYNONYMS: Table 4 SAMPLING MEASUREMENT SAMPLER: FILTER/SOLID SORBENT TUBE (OVS-2 tube: TECHNIQUE: GC, FLAME PHOTOMETRIC DETECTION 13-mm quartz filter; XAD-2, 270 mg/140 mg) (FPD) FLOW RATE: 0.2 to 1 L/min ANALYTE: organophosphorus pesticides, Table 1 VOL-MIN: 12 L EXTRACTION: 2-mL 90% toluene/10% acetone solution -MAX: 240 L; 60 L (Malathion, Ronnel) INJECTION SHIPMENT: cap both ends of tube VOLUME: 1-2 µL SAMPLE TEMPERATURE STABILITY: at least 10 days at 25 °C -INJECTION: 240 °C at least 30 days at 0 °C -DETECTOR: 180 °C to 215 °C (follow manufacturer's recommendation) BLANKS: 2 to 10 field blanks per set -COLUMN: Table 6 CARRIER GAS: He at 15 psi (104 kPa) ACCURACY COLUMN: fused silica capillary column; Table 6 RANGE STUDIED: Table 5, Column A DETECTOR: FPD (phosphorus mode) ACCURACY: Table 5, Column B CALIBRATION: standard solutions of organophosphorus compounds in toluene BIAS: Table 5, Column C RANGE: Table 8, Column C ˆ OVERALL PRECISION (S rT): Table 5, Column D ESTIMATED LOD: Table 8, Column F PRECISION (S r): Table 5, Column E APPLICABILITY: The working ranges are listed in Table 5. They cover a range of 1/10 to 2 times the OSHA PELs. This INTERFERENCES: Several organophosphates may co-elute method also is applicable to STEL measurements using 12-L with either target analyte or internal standard causing samples. -

Parathion-Methyl
FAO SPECIFICATIONS AND EVALUATIONS FOR PLANT PROTECTION PRODUCTS PARATHION-METHYL O,O-dimethyl O-4-nitrophenyl phosphorothioate 2001 TABLE OF CONTENTS PARATHION-METHYL Page DISCLAIMER 3 INTRODUCTION 4 PART ONE 5 SPECIFICATIONS FOR PARATHION-METHYL PARATHION-METHYL INFORMATIONERROR! BOOKMARK NOT DEFINED. PARATHION-METHYL TECHNICAL MATERIAL 6 PARATHION-METHYL TECHNICAL CONCENTRATE 8 PARATHION-METHYL EMULSIFIABLE CONCENTRATE 10 PART TWO 13 2001 EVALUATION REPORT ON PARATHION-METHYL 14 Page 2 of 31 PARATHION-METHYL SPECIFICATIONS 2001 Disclaimer1 FAO specifications are developed with the basic objective of ensuring that pesticides complying with them are satisfactory for the purpose for which they are intended so that they may serve as an international point of reference. The specifications do not constitute an endorsement or warranty of the use of a particular pesticide for a particular purpose. Neither do they constitute a warranty that pesticides complying with these specifications are suitable for the control of any given pest, or for use in a particular area. Owing to the complexity of the problems involved, the suitability of pesticides for a particular application must be decided at the national or provincial level. Furthermore, the preparation and use of pesticides complying with these specifications are not exempted from any safety regulation or other legal or administrative provision applicable thereto. FAO shall not be liable for any injury, loss, damage or prejudice of any kind that may be suffered as a result of the preparation, transportation, sale or use of pesticides complying with these specifications. Additionally, FAO wishes to alert users of specifications to the fact that improper field mixing and/or application of pesticides can result in either a lowering or complete loss of efficacy. -

Reactivity)And)Continued)Activity)Of)Immobilized)Zinc)
! ! REACTIVITY)AND)CONTINUED)ACTIVITY)OF)IMMOBILIZED)ZINC) OXIDE)NANOPARTICLES)ON)METHYL)PARATHION) DECONTAMINATION) ! ! ! A!Thesis! Presented!to!the!Faculty!of!the!Graduate!School! of!Cornell!University! In!Partial!Fulfillment!of!the!Requirements!for!the!Degree!of! Master!of!Science! ! ! ! ! By!Yunfei!Han! May!2014! ) ) ! ! ! ! ! ! ! ! ! ©!Yunfei!Han!2014! ALL!RIGHTS!RESERVED! ! ! ! ! ! ABSTRACT) Photocatalytic! degradation! activity! and! reusability! of! ZnO! nanoparticles! and! immobilized! ZnO! in! polyacrylonitrile! (PAN)! nanofibers! were! investigated.! It! was! found! that! photocatalytic! degradation! using! ZnO! is! a! rapid! and! effective! way! to! degrade! methyl! parathion.! The! photocatalytic! degradation! reactivity! of! ZnO! nanoparticles! remained! in! four! cycles! with! some! loss! of! activity,! while! the! photocatalytic!degradation!reactivity!of!ZnO/PAN!nanofibers!remained!in!five!cycles! with!no!loss!of!activity!detected.!By!analyzing!samples!using!HPLC!chromatograms,! mass!spectra,!UVXvis!spectra,!calculated!octanolXwater!partition!coefficients,!and!31P! NMR! spectrum,! it! was! shown! that! the! possible! mechanism! of! the! photocatalytic! degradation!in!water/ethanol!is!predominated!by!hydrolysis.! ! iii! ! BIOGRAPHICAL)SKETCH) Yunfei!Han!was!born!and!raised!in!Shanghai,!P.R.!China.!Before!beginning!her! M.S.!study!in!Fiber!Science!program!at!Cornell!University,!she!earned!a!B.E.!degree! in!the!field!of!Textile!Science!and!Engineering!from!Donghua!University,!Shanghai,!P.! R.!China.!During!her!undergraduate!study,!she!spent!her!senior!year!in!UC!Davis!as! -

Recent Advances on Detection of Insecticides Using Optical Sensors
sensors Review Recent Advances on Detection of Insecticides Using Optical Sensors Nurul Illya Muhamad Fauzi 1, Yap Wing Fen 1,2,*, Nur Alia Sheh Omar 1,2 and Hazwani Suhaila Hashim 2 1 Functional Devices Laboratory, Institute of Advanced Technology, Universiti Putra Malaysia, Serdang 43400, Selangor, Malaysia; [email protected] (N.I.M.F.); [email protected] (N.A.S.O.) 2 Department of Physics, Faculty of Science, Universiti Putra Malaysia, Serdang 43400, Selangor, Malaysia; [email protected] * Correspondence: [email protected] Abstract: Insecticides are enormously important to industry requirements and market demands in agriculture. Despite their usefulness, these insecticides can pose a dangerous risk to the safety of food, environment and all living things through various mechanisms of action. Concern about the environmental impact of repeated use of insecticides has prompted many researchers to develop rapid, economical, uncomplicated and user-friendly analytical method for the detection of insecticides. In this regards, optical sensors are considered as favorable methods for insecticides analysis because of their special features including rapid detection time, low cost, easy to use and high selectivity and sensitivity. In this review, current progresses of incorporation between recognition elements and optical sensors for insecticide detection are discussed and evaluated well, by categorizing it based on insecticide chemical classes, including the range of detection and limit of detection. Additionally, this review aims to provide powerful insights to researchers for the future development of optical sensors in the detection of insecticides. Citation: Fauzi, N.I.M.; Fen, Y.W.; Omar, N.A.S.; Hashim, H.S. Recent Keywords: insecticides; optical sensor; recognition element Advances on Detection of Insecticides Using Optical Sensors. -

High Hazard Chemical Policy
Environmental Health & Safety Policy Manual Issue Date: 2/23/2011 Policy # EHS-200.09 High Hazard Chemical Policy 1.0 PURPOSE: To minimize hazardous exposures to high hazard chemicals which include select carcinogens, reproductive/developmental toxins, chemicals that have a high degree of toxicity. 2.0 SCOPE: The procedures provide guidance to all LSUHSC personnel who work with high hazard chemicals. 3.0 REPONSIBILITIES: 3.1 Environmental Health and Safety (EH&S) shall: • Provide technical assistance with the proper handling and safe disposal of high hazard chemicals. • Maintain a list of high hazard chemicals used at LSUHSC, see Appendix A. • Conduct exposure assessments and evaluate exposure control measures as necessary. Maintain employee exposure records. • Provide emergency response for chemical spills. 3.2 Principle Investigator (PI) /Supervisor shall: • Develop and implement a laboratory specific standard operation plan for high hazard chemical use per OSHA 29CFR 1910.1450 (e)(3)(i); Occupational Exposure to Hazardous Chemicals in Laboratories. • Notify EH&S of the addition of a high hazard chemical not previously used in the laboratory. • Ensure personnel are trained on specific chemical hazards present in the lab. • Maintain Material Safety Data Sheets (MSDS) for all chemicals, either on the computer hard drive or in hard copy. • Coordinate the provision of medical examinations, exposure monitoring and recordkeeping as required. 3.3 Employees: • Complete all necessary training before performing any work. • Observe all safety -
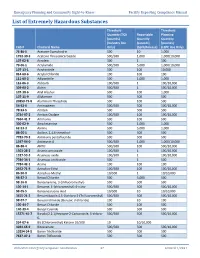
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

Insecticides
fY)I\) -;; ooo 3tfJ INSECTICIDES Extension Bulletin 387-Revlsed 1980 AGAfCULTURAL EXTENSION SERVICE UNIVERSITY OF MINNESOTA Contents General precautions for using pesticides . 4 Safety precautions and first aid . 4 Minnesota poison information centers . 5 Protecting honey bees from insecticides . 6 Pesticide toxicity and LD 50's . • . • . 6 Acute oral and dermal LD 50's for insecticides . 7 Forms of insecticides . 8 Calculating dosage and rates of application . 9 Sprayer calibration . 11 Description of insecticides, miticides ........................... 12 Chlorinated hydrocarbons ................................. 12 Carbamates .............................................. 13 Organophosphates ........................................ 14 Sulfonates, carbonates, botanicals, and miscellaneous groups ... 18 Legal Restrictions on Use of Pesticides The Federal Insecticide, Fungicide and Rodenticide Act and the Minnesota Pesticide Act of 1976, require that those who use or supervise the use of certain pesticides with restricted uses must be certified. The labels of those pesticides with restricted uses will contain information regarding these restrictions. Be sure to read all labels thoroughly and use any pesticide for the crops and pests listed on the label only. Information about applicator certification may be obtained from your County Extension Director or the Minnesota Depmt ment of Agriculture. The U.S. Environmental Protection Agency (EPA) has designated the following pesticides for reshicted use: acrolein endrin mevinphos (Phosdrin) acrylonitrile ethyl parathion paraquat aldicarb (Temik) 1080 piclorarri (Tordon) allyl alcohol 1081 sodium cyanide alluminum phosphide (Phostoxin) hydrocyanic acid strychnine azinphos methyl (Guthion) methomyl (Lannate, Nudrin) sulfotepp calcium cyanide methyl bromide tepp demeton (Systox) methyl parathion Authors of this publication are J. A. Lofgren, professor and extension entomologist; D. M. Noetzel, assistant professor and extension entomologist; P. K. Hareln, professor and extension entomologist; M. -

Spider Mites and Resistance 653 in Production and Better Flowers V/Ith Quired Dosages
day to nearly 2 days. Then it enters the resting stage. After a day or so it molts to a second active stage, which feeds Spider Mites and and then becomes quiescent as in the first stage. The adult male emerges Resistance from the second quiescent stage, but the female passes through a third feed- Floyd F. Smith ing and quiescent stage before becom- ing an adult. Mating usually occurs within a few minutes after the female Spider mites, or "red spiders," at- becomes an adult. Only males develop tack nearly all kinds of field crops, veg- from eggs of unmated females. etables, orchard trees, weeds, and The developmental period varies greenhouse plants. The mites often v-^-idely with the temperature. The eggs confine themselves to one kind of plant hatch in 2 or 3 days at 75° F. or higher at first but move to other kinds when or after 21 days at 55°. The mites may injury increases and food becomes reach the adult stage within 5 days at scarce. 75° or in 40 days at 55°. Under aver- Several species, of similar habits but age greenhouse temperatures of 60° to different morphological characters, arc 70°, the incubation period is 5 to 10 found out of doors. days and development to adult stage Of six species that infest green- from 10 to 15 days. One female lays a houses, the two-spotted spider mite is few eggs daily and a total of 100 to predominant. It is a general feeder 194 eggs during an average life of 3 to but it is almost constantly present on 4 weeks. -

Methyl Parathion 19
METHYL PARATHION 19 3. HEALTH EFFECTS 3.1 INTRODUCTION The primary purpose of this chapter is to provide public health officials, physicians, toxicologists, and other interested individuals and groups with an overall perspective on the toxicology of methyl parathion. It contains descriptions and evaluations of toxicological studies and epidemiological investigations and provides conclusions, where possible, on the relevance of toxicity and toxicokinetic data to public health. A glossary and list of acronyms, abbreviations, and symbols can be found at the end of this profile. 3.2 DISCUSSION OF HEALTH EFFECTS BY ROUTE OF EXPOSURE To help public health professionals and others address the needs of persons living or working near hazardous waste sites, the information in this section is organized first by route of exposure (inhalation, oral, and dermal) and then by health effect (death, systemic, immunological, neurological, reproductive, developmental, genotoxic, and carcinogenic effects). These data are discussed in terms of three exposure periods: acute (14 days or less), intermediate (15–364 days), and chronic (365 days or more). Levels of significant exposure for each route and duration are presented in tables and illustrated in figures. The points in the figures showing no-observed-adverse-effect levels (NOAELs) or lowest-observed-adverse-effect levels (LOAELs) reflect the actual doses (levels of exposure) used in the studies. LOAELS have been classified into "less serious" or "serious" effects. "Serious" effects are those that evoke failure in a biological system and can lead to morbidity or mortality (e.g., acute respiratory distress or death). "Less serious" effects are those that are not expected to cause significant dysfunction or death, or those whose significance to the organism is not entirely clear. -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals