A THESIS Entitled the ADDITION of HALOGEN
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Use of Solvents for Pahs Extraction and Enhancement of the Pahs Bioremediation in Coal- Tar-Contaminated Soils Pak-Hing Lee Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2000 Use of solvents for PAHs extraction and enhancement of the PAHs bioremediation in coal- tar-contaminated soils Pak-Hing Lee Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Environmental Engineering Commons Recommended Citation Lee, Pak-Hing, "Use of solvents for PAHs extraction and enhancement of the PAHs bioremediation in coal-tar-contaminated soils " (2000). Retrospective Theses and Dissertations. 13912. https://lib.dr.iastate.edu/rtd/13912 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter fece, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quaiity of the copy submitted. Broken or indistinct print colored or poor quality illustrations and photographs, print bleedthrough, substeindard margins, and improper alignment can adversely affect reproduction. In the unlilcely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -
Chapter 13.Pptx
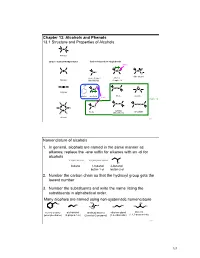
Chapter 13: Alcohols and Phenols 13.1 Structure and Properties of Alcohols C C Alkanes Carbon - Carbon Multiple Bonds Carbon-heteroatom single bonds basic O C C C N C N C X O nitro alkane X= F, Cl, Br, I amines Alkenes Alkyl Halide Chapter 23 OH C C H O C O C C O C C Alkynes phenol alcohols ethers epoxide acidic Chapter 14 H H H C S C C C C S S C C S C C H C C sulfides thiols disulfide H H (thioethers) Arenes 253 Nomenclature of alcohols 1. In general, alcohols are named in the same manner as alkanes; replace the -ane suffix for alkanes with an -ol for alcohols CH3CH2CH2CH3 CH3CH2CH2CH2OH OH butane 1-butanol 2-butanol butan-1-ol butan-2-ol 2. Number the carbon chain so that the hydroxyl group gets the lowest number 3. Number the substituents and write the name listing the substituents in alphabetical order. Many alcohols are named using non-systematic nomenclature H C OH 3 OH OH C OH OH HO OH H3C HO H3C benzyl alcohol allyl alcohol tert-butyl alcohol ethylene glycol glycerol (phenylmethanol) (2-propen-1-ol) (2-methyl-2-propanol) (1,2-ethanediol) (1,2,3-propanetriol) 254 127 Alcohols are classified according to the H R C OH C OH H H degree of substitution of the carbon bearing H H 1° carbon the -OH group methanol primary alcohol primary (1°) : one alkyl substituent R R C OH C OH R R secondary (2°) : two alkyl substituents H R 2° carbon 3° carbon tertiary (3°) : three alkyl substituents secondary alcohol tertiary alcohol Physical properties of alcohols – the C-OH bond of alcohols has a significant dipole moment. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Supporting Information of Polycyclic Aromatic Hydrocarbons (Pahs) In
Supporting Information of Polycyclic aromatic hydrocarbons (PAHs) in aerosols over the central Himalayas along two south-north transects Peng Fei Chen1,5, Chao Liu Li1, Shi Chang Kang2,3*, Maheswar Rupakheti4, Arnico K Panday6, Fang Ping Yan2,5, Quan Lian Li2, Qiang Gong Zhang1,3, Jun Ming Guo1,5, Dipesh Rupakheti1,5, Wei Luo7 1Key Laboratory of Tibetan Environment Changes and Land Surface Processes, Institute of Tibetan Plateau Research, Chinese Academy of Sciences, Beijing 100101, China 2State Key Laboratory of Cryospheric Science, Cold and Arid Regions Environmental and Engineering Research Institute, Lanzhou 730000, China 3Center for Excellence in Tibetan Plateau Earth Sciences, Chinese Academy of Sciences, Beijing 100085, China; 4Institute for Advanced Sustainability Studies, Potsdam 14467, Germany 5University of Chinese Academy of Sciences, Beijing 100039, China 6International Centre for Integrated Mountain Development, Kathmandu, Nepal 7State Key Lab of Urban and Regional Ecology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China Correspondence to: S. Kang ([email protected]) Text SI-1 Sample extraction and analysis Text SI-2 Quality control Table SI-1 Percentage contribution (%) of each species to total PAHs in the atmosphere over the Himalayas. Text SI-1 Sample extraction and analysis A quarter of each filter was cut into pieces, placed into a glass tube, and immersed in 20 mL of dichloromethane (DCM) and n-hexane (1:1). The extraction was performed by sonication twice for 30 min at 27 °C. Every single sample was spiked with deuterated PAHs (naphthalene-d8, acenaphthene-d10, phenanthrene-d10, chrysene-d12, and perylene-d12) as recovery surrogates. -

Reductions and Reducing Agents
REDUCTIONS AND REDUCING AGENTS 1 Reductions and Reducing Agents • Basic definition of reduction: Addition of hydrogen or removal of oxygen • Addition of electrons 9:45 AM 2 Reducible Functional Groups 9:45 AM 3 Categories of Common Reducing Agents 9:45 AM 4 Relative Reactivity of Nucleophiles at the Reducible Functional Groups In the absence of any secondary interactions, the carbonyl compounds exhibit the following order of reactivity at the carbonyl This order may however be reversed in the presence of unique secondary interactions inherent in the molecule; interactions that may 9:45 AM be activated by some property of the reacting partner 5 Common Reducing Agents (Borohydrides) Reduction of Amides to Amines 9:45 AM 6 Common Reducing Agents (Borohydrides) Reduction of Carboxylic Acids to Primary Alcohols O 3 R CO2H + BH3 R O B + 3 H 3 2 Acyloxyborane 9:45 AM 7 Common Reducing Agents (Sodium Borohydride) The reductions with NaBH4 are commonly carried out in EtOH (Serving as a protic solvent) Note that nucleophilic attack occurs from the least hindered face of the 8 carbonyl Common Reducing Agents (Lithium Borohydride) The reductions with LiBH4 are commonly carried out in THF or ether Note that nucleophilic attack occurs from the least hindered face of the 9:45 AM 9 carbonyl. Common Reducing Agents (Borohydrides) The Influence of Metal Cations on Reactivity As a result of the differences in reactivity between sodium borohydride and lithium borohydride, chemoselectivity of reduction can be achieved by a judicious choice of reducing agent. 9:45 AM 10 Common Reducing Agents (Sodium Cyanoborohydride) 9:45 AM 11 Common Reducing Agents (Reductive Amination with Sodium Cyanoborohydride) 9:45 AM 12 Lithium Aluminium Hydride Lithium aluminiumhydride reacts the same way as lithium borohydride. -

UNIT 6 ELEMENTS of GROUP 13 Structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction Uses I 6.3 General Characteristics
UNIT 6 ELEMENTS OF GROUP 13 structure 6.1 Introduction Objectives I 6.2 Oailcrence, Extraction and Uses Occurrec - Extraction uses i 6.3 General Characteristics I - 6.5 Halides of Bpron anel Aluminium Halides of Boron Halides of Aluminium 6.6 Oxides of Boron and Aluminium I Boric Oxide Aluminium Oxide 6.7 Oxoacids of Boron and Borates 6.8 Borazine 6.9 Complexation Behaviour 6.10 Anomalous Behaviour of Boron 6.11 Summary 6.12 Terminal Questions 6.13- Answers ' -- 6.1 INTRODUCTION In the previous two units, you studied the main features of the chemistry of Group 1 and Group 2 elements, i.e. the alkali and the alkaline earth metals. In this unit you - will study the elements of Group 13, namely, boron, aluminium, gallium, indium and, thallium. While studying the alkali and alkaline earth metals, you have seen that all Zhe elements of these two groups are highly reactive metals and the first element of each group shows some differences from the rest. In Group 13, the differences between the first element and the remaining elements become so pronounced that the first member of the group, i.e. boron is a nonmetal wheieas the rest of the elements are distinctly metallic in nature. In a way, this is the first group of the periodic table in which you observe a marked change in the hature of the elements . down the group. describe the chemistry of hydrides, halides and oxides of boron and aluminium, elucidate the structures of hydrides of boron and aluminium, 6.2 OCCURRENC~,EXTRACTION AND USES Elements bf Group 13 are sufficiently reactive. -

Hydrogen Transmutation of Nickel in Glow Discharge
International Journal of Materials Science ISSN 0973-4589 Volume 12, Number 3 (2017), pp. 405-409 © Research India Publications http://www.ripublication.com Hydrogen Transmutation of Nickel in Glow Discharge Vladimir K. Nevolin National Research University of Electronic Technology (MIET), Moscow, Russia. Abstract Background: The possibility of the existence of subatomic hydrogen states was theoretically predicted previously. Objectives: Prove that the transmutation of elements is possible in specially prepared conditions for hydrogen. Methodology: By comparing the mass spectra of deposits on silicon substrates and target electrodes, it is shown that a change in the composition is observed in a magnetron Argon. Results: An increase in the concentration of 62 60 the nickel isotope 28 Ni and a decrease in the isotope concentration 28 Ni are shown. Conclusion: These results confirm the results obtained earlier in the heat generator Rossi, who worked more than a year, found an increase in the 62 isotope 28 Ni due to a decrease in the proportion of other isotopes. Keywords: transmutation, isotopes of nickel, glow discharge, argon, hydrogen INTRODUCION It is considered that the cold transmutation of elements (cold nuclear reactions) has been experimentally demonstrated [1]. On the basis of this phenomenon, energy generators are created in which long-term release of thermal energy in excess of expended energy is observed [2]. From many experimental studies it can be seen that hydrogen, which plays a pivotal role in the reaction zone, may be delivered through a variety of chemical compounds; for example, using lithium aluminium hydride LiAlH4. An analysis of the products of nuclear reactions suggests the possibility of many simultaneous nuclear fusion and decomposition reactions [3]. -

Effect of Epoxide Content on the Vulcanizate Structure of Silica-Filled Epoxidized Natural Rubber (ENR) Compounds
polymers Article Effect of Epoxide Content on the Vulcanizate Structure of Silica-Filled Epoxidized Natural Rubber (ENR) Compounds Gyeongchan Ryu 1, Donghyuk Kim 1, Sanghoon Song 1, Kiwon Hwang 1, Byungkyu Ahn 2 and Wonho Kim 1,* 1 School of Chemical Engineering, Pusan National University, Busan 46241, Korea; [email protected] (G.R.); [email protected] (D.K.); [email protected] (S.S.); [email protected] (K.H.) 2 Hankook Tire & Technology Co., Ltd., R&D Center, 50 Yuseong-daero 935 beon-gil, Yuseong-gu, Daejeon 34127, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-51-510-3190 Abstract: The demand for truck–bus radial (TBR) tires with enhanced fuel efficiency has grown in recent years. Many studies have investigated silica-filled natural rubber (NR) compounds to address these needs. However, silica-filled compounds offer inferior abrasion resistance compared to carbon black-filled compounds. Further, the use of NR as a base rubber can hinder silanization and coupling reactions due to interference by proteins and lipids. Improved silica dispersion be achieved without the use of a silane coupling agent by introducing epoxide groups to NR, which serve as silica-affinitive functional groups. Furthermore, the coupling reaction can be promoted by facilitating chemical interaction between the hydroxyl group of silica and the added epoxide groups. Thus, this study evaluated the properties of commercialized NR, ENR-25, and ENR-50 compounds with or without an added silane coupling agent, and the filler–rubber interaction was quantitatively calculated using vulcanizate structure analysis. The increased epoxide content, when the silane coupling agent was not used, improved silica dispersion, abrasion resistance, fuel efficiency, and wet Citation: Ryu, G.; Kim, D.; Song, S.; grip. -

Organic Chemistry Name Formula Isomers Methane CH 1 Ethane C H
Organic Chemistry Organic chemistry is the chemistry of carbon. The simplest carbon molecules are compounds of just carbon and hydrogen, hydrocarbons. We name the compounds based on the length of the longest carbon chain. We then add prefixes and suffixes to describe the types of bonds and any add-ons the molecule has. When the molecule has just single bonds we use the -ane suffix. Name Formula Isomers Methane CH4 1 Ethane C2H6 1 Propane C3H8 1 Butane C4H10 2 Pentane C5H12 3 Hexane C6H14 5 Heptane C7H16 9 Octane C8H18 18 Nonane C9H20 35 Decane C10H22 75 Isomers are compounds that have the same formula but different bonding. isobutane n-butane 1 Naming Alkanes Hydrocarbons are always named based on the longest carbon chain. When an alkane is a substituent group they are named using the -yl ending instead of the -ane ending. So, -CH3 would be a methyl group. The substituent groups are named by numbering the carbons on the longest chain so that the first branching gets the lowest number possible. The substituents are listed alphabetically with out regard to the number prefixes that might be used. 3-methylhexane 1 2 3 4 5 6 6 5 4 3 2 1 Alkenes and Alkynes When a hydrocarbon has a double bond we replace the -ane ending with -ene. When the hydrocarbon has more than three carbon the position of the double bond must be specified with a number. 1-butene 2-butene Hydrocarbons with triple bonds are named basically the same, we replace the -ane ending with -yne. -

WO 2015/177807 Al 26 November 2015 (26.11.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/177807 Al 26 November 2015 (26.11.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every C07D 403/14 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/IN20 15/000066 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, 3 February 2015 (03.02.2015) KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (25) Filing Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (26) Publication Language: English SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 1709/MUM/2014 22 May 2014 (22.05.2014) (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: WANBURY LTD. [IN/IN]; BSEL tech park, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, B wing, 10th floor, sector 30A, opp. -

Stability Studies of Selected Polycyclic Aromatic Hydrocarbons in Different Organic Solvents and Identification of Their Transformation Products
Polish J. of Environ. Stud. Vol. 17, No. 1 (2008), 17-24 Original Research Stability Studies of Selected Polycyclic Aromatic Hydrocarbons in Different Organic Solvents and Identification of Their Transformation Products D. Dąbrowska1, A. Kot-Wasik2, J. Namieśnik*2 1Polish geological institute, Central Chemical laboratory, warsaw, Poland 2Department of Analytical Chemistry, gdansk university of technology, ul. Narutowicza 11/12, 80-892 gdańsk, Poland Received: May 7, 2007 Accepted: October 1, 2007 Abstract one of the problems in an hPlC laboratory is the preservation of samples and extracts prior to analysis without any changes. It has been found that degradation processes cannot be eliminated entirely, but they can be slowed down considerably. Photodegradation kinetics of fluorene, anthracene and benzo(a)pyrene were studied in various organic solvents. Solvents known as good media to store PAhs for a long time were selected with respect to avoid degradation. in the case of fluorene, 9-fluorenone and 9-hydroxyfluorene were detected as main photoproducts. Formation of anthraquinone and 1,8-dihydroxyanthraquinone during the degradation of anthracene was observed. Benzo(a)pyrene-4,5-dihydrodiol and one of the isomers of hydroxy-BaP-dione as products of benzo(a)pyrene photodegradation have been identified. Keywords: polycyclic aromatic hydrocarbons, photodegradation, degradation products, sample stability Introduction 4 or more rings and their metabolites, have a variety of mutagenic and carcinogenic effects on microorganisms, Polycyclic aromatic hydrocarbons (PAhs) are ubiq- plants and animals, and are classified as compounds with uitous contaminants originating from natural and anthro- significant human health risks [6]. pogenic pyrolysis of organic matter such as forest fires, In the environment, primary removal processes of low automobile exhaust, coal and oil refining processes.t heir molecular weight PAhs are microbial degradation and abundance and persistence in several polluted environ- evaporation. -

Lithium Aluminium Hydride.Pdf
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 5.0 Revision Date 03/12/2011 Print Date 09/12/2011 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Aluminium lithium hydride Product Number : 531502 Brand : Aldrich Supplier : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # (For : (314) 776-6555 both supplier and manufacturer) Preparation Information : Sigma-Aldrich Corporation Product Safety - Americas Region 1-800-521-8956 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Water Reactive, Target Organ Effect, Toxic by ingestion, Corrosive Target Organs Kidney, Liver, Central nervous system GHS Classification Substances, which in contact with water, emit flammable gases (Category 1) Acute toxicity, Oral (Category 3) Skin corrosion (Category 1B) Serious eye damage (Category 1) GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H260 In contact with water releases flammable gases which may ignite spontaneously. H301 Toxic if swallowed. H314 Causes severe skin burns and eye damage. Precautionary statement(s) P223 Keep away from any possible contact with water, because of violent reaction and possible flash fire. P231 + P232 Handle under inert gas. Protect from moisture. P280 Wear protective gloves/ protective clothing/ eye protection/ face protection. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P310 Immediately call a POISON CENTER or doctor/ physician. P370 + P378 In case of fire: Use dry sand, dry chemical or alcohol-resistant foam for extinction. P422 Store contents under inert gas.