Indium Arsenide/Gallium Arsenide Quantum Dots and Nanomesas: Multimillion-Atom Molecular Dynamics Solutions on Parallel Architectures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TR-499: Indium Phosphide (CASRN 22398-80-7) in F344/N Rats And
NTP TECHNICAL REPORT ON THE TOXICOLOGY AND CARCINOGENESIS STUDIES OF INDIUM PHOSPHIDE (CAS NO. 22398-80-7) IN F344/N RATS AND B6C3F1 MICE (INHALATION STUDIES) NATIONAL TOXICOLOGY PROGRAM P.O. Box 12233 Research Triangle Park, NC 27709 July 2001 NTP TR 499 NIH Publication No. 01-4433 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service National Institutes of Health FOREWORD The National Toxicology Program (NTP) is made up of four charter agencies of the U.S. Department of Health and Human Services (DHHS): the National Cancer Institute (NCI), National Institutes of Health; the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health; the National Center for Toxicological Research (NCTR), Food and Drug Administration; and the National Institute for Occupational Safety and Health (NIOSH), Centers for Disease Control and Prevention. In July 1981, the Carcinogenesis Bioassay Testing Program, NCI, was transferred to the NIEHS. The NTP coordinates the relevant programs, staff, and resources from these Public Health Service agencies relating to basic and applied research and to biological assay development and validation. The NTP develops, evaluates, and disseminates scientific information about potentially toxic and hazardous chemicals. This knowledge is used for protecting the health of the American people and for the primary prevention of disease. The studies described in this Technical Report were performed under the direction of the NIEHS and were conducted in compliance with NTP laboratory health and safety requirements and must meet or exceed all applicable federal, state, and local health and safety regulations. Animal care and use were in accordance with the Public Health Service Policy on Humane Care and Use of Animals. -

Advanced VCSEL Technology: Self-Heating and Intrinsic Modulation Response
IEEE JOURNAL OF QUANTUM ELECTRONICS, VOL. 54, NO. 3, JUNE 2018 2400209 Advanced VCSEL Technology: Self-Heating and Intrinsic Modulation Response Dennis G. Deppe, Fellow, IEEE, Mingxin Li, Xu Yang, and Mina Bayat (Invited Paper) Abstract— Experimental data and modeling results are can efficiently conduct heat from the strongly pumped laser presented for a new type of vertical-cavity surface-emitting active volume, and in a laser cavity much smaller than possible laser (VCSEL) that solves numerous problems with oxide before with commercial edge-emitting laser diodes. VCSELs. In addition, the new oxide-free VCSEL can be scaled to small size. Modeling shows that this small size can dramatically This change made it possible to obtain much higher cur- increase the speed of the laser through control of self-heating. rent density, and therefore higher photon density, and higher Modeling compared with both the oxide and the new oxide-free stimulated emission rate than other laser diodes. Continually VCSEL predict intrinsic modulation speed for room temperature reducing the laser active volume and relying on relatively > approaching 80 GHz, indicating the potential for 100-Gb/s data high efficiency VCSEL designs have produced ever-increasing speed from directly modulated VCSELs. The modeling is one of if not the first to include self-heating and spectral detuning between laser diode speed. VCSELs now reign supreme in laboratory the gain and the cavity resonance that results from self-heating. demonstrations of direct modulation data speed due largely to The modeling results accurately accounts for the saturation in their combined small volume, matched cavity and gain region, modulation speed in very high-speed oxide VCSELs operating at and high internal heat flow through high quality epitaxial high temperature and accounts for the temperature of differential mirrors [15]–[23]. -

Scalable Synthesis of Inas Quantum Dots Mediated Through Indium Redox Chemistry Matthias Ginterseder, Daniel Franke, Collin F
pubs.acs.org/JACS Communication Scalable Synthesis of InAs Quantum Dots Mediated through Indium Redox Chemistry Matthias Ginterseder, Daniel Franke, Collin F. Perkinson, Lili Wang, Eric C. Hansen, and Moungi G. Bawendi* Cite This: J. Am. Chem. Soc. 2020, 142, 4088−4092 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: Next-generation optoelectronic applications centered in the near-infrared (NIR) and short-wave infrared (SWIR) wavelength regimes require high-quality materials. Among these materials, colloidal InAs quantum dots (QDs) stand out as an infrared-active candidate material for biological imaging, lighting, and sensing applications. Despite significant development of their optical properties, the synthesis of InAs QDs still routinely relies on hazardous, commercially unavailable precursors. Herein, we describe a straightforward single hot injection procedure revolving around In(I)Cl as the key precursor. Acting as a simultaneous reducing agent and In source, In(I)Cl smoothly reacts with a tris(amino)arsenic precursor to yield colloidal InAs quantitatively and at gram scale. Tuning the reaction temperature produces InAs cores with a first excitonic absorption feature in the range of 700− 1400 nm. A dynamic disproportionation equilibrium between In(I), In metal, and In(III) opens up additional flexibility in precursor selection. CdSe shell growth on the produced cores enhances their optical properties, furnishing particles with center emission wavelengths between 1000 and 1500 nm and narrow photoluminescence full-width at half-maximum (FWHM) of about 120 meV throughout. The simplicity, scalability, and tunability of the disclosed precursor platform are anticipated to inspire further research on In-based colloidal QDs. -

III-V Material Properties Welcome to This Section on III-V PV Technology. in This Video We Will Discuss the Material
III-V Material Properties Welcome to this section on III-V PV technology. In this video we will discuss the material properties of the semiconductors used for this technology. In this video, you will learn what the III-V PV technology exactly is and how it is different from other solar cell technologies. We will then discuss the structural properties of III-V semiconductors. In the next video we will look into the electrical and optical properties of III- V materials. The III-V PV technology, as the name implies, utilizes elements of the 3-A group and the 5-A group in the periodic table of elements. As you might recall, group 3 elements have 3 electrons in their outer valence shell, and group 5 elements have 5 valence electrons. One of the most common III-V absorber materials is formed by bonding the group 3 material gallium, with the group 5 material arsenic. Several such combinations are possible however, and many different III-V alloys are used as an absorber material by the PV industry. Examples include Gallium phosphide, Indium phosphide, indium arsenide, Gallium indium arsenide, gallium indium phosphide, aluminium-gallium-indium-arsenide and aluminium- gallium-indium-phosphide. An important question for III-V technology is how abundant are the III- and V- elements. As discussed at the start of this course, of these five elements, only aluminium and phosphorus are relatively abundant in nature. Gallium, arsenide and germanium are not rare or precious metals, but they are much less abundant than silicon. Indium is rare and in high demand. -

Environmental Protection Agency § 712.30
Environmental Protection Agency § 712.30 (2) As a non-isolated intermediate. ment Information Manufacturer’s Re- (3) As an impurity. port for each chemical substance or [47 FR 26998, June 22, 1982; 47 FR 28382, June mixture that is listed or designated in 30, 1982] this section. (2) Unless a respondent has already § 712.28 Form and instructions. prepared a Manufacturer’s Report in (a) Manufacturers and importers sub- conformity with conditions set forth in ject to this subpart must submit a sin- paragraph (a)(3) of this section, the in- gle EPA Form No. 7710–35, ‘‘Manufac- formation in each Manufacturer’s Re- turer’s Report—Preliminary Assess- port must cover the respondent’s latest ment Information,’’ for each plant site complete corporate fiscal year as of the manufacturing or importing a chem- effective date. The effective date will ical substance listed in § 712.30. be 30 days after the FEDERAL REGISTER (b) Reporting companies may submit publishes a rule amendment making their reports through individual plant the substance or mixture subject to sites or company headquarters as they this subpart B. choose. A separate form must be sub- (3) Persons subject to this subpart B mitted for each plant site manufac- need not comply with the requirements turing the chemical substance. of paragraph (a)(2) of this section if (c) You must submit forms by one of they meet either one of the following the following methods: conditions: (1) Mail, preferably certified, to the (i) The respondent has previously and Document Control Office (DCO) voluntarily provided EPA with a Manu- (7407M), Office of Pollution Prevention facturer’s Report on a chemical sub- and Toxics (OPPT), Environmental stance or mixture subject to this sub- Protection Agency, 1200 Pennsylvania part B, which contains data for a one- Ave., NW., Washington, DC 20460–0001, year period ending no more than three ATTN: 8(a) PAIR Reporting. -

Properties of Indium-Arsenide Measured by Low Temperature Photoluminescence
PROPERTIES OF INDIUM-ARSENIDE MEASURED BY LOW TEMPERATURE PHOTOLUMINESCENCE YVESLACROIX B.Sc.(Honours) S.Sp. Physics, Queen's University, 1991 THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in the Department of PHYSICS @ Yves Lacroix 1996 SIMON FRASER UNIVERSITY April, 1996 All rights reserved. This work may not be reproduced in whole or in part, by photocopy or other means, without permission of the author. Approval Page NAME: Yves Lacroix DEGREE: Doctor of Philosophy (Physics) TITLE OF THESIS: Properties of Indium-Arsenide Measured by Low Temperature Photoluminescence EXAMINING COMMITTEE: CHAIR: Jeff Dahn Mike Thewalt Senior Supervisor Professor of Physics Simon Watkins Assistant Professor of Physics ~ohn-echhoefer ' Internal External Examiner Professor of Physics Alain Roth External Examiner Senior research officer, C.N.R.C. PARTLAL COPYRIGHT LICENSE I hereby grant to Simon Fraser Universi the right to lend my thesis, pro'ect or extended essay (the title o7 which is shown below) to users od the Simon Fraser University Library, and to make partial or single copies only for such users or in response to a request from the library of any other university, or other educational institution, on its own behalf or for one of its users. I further agree that permission for multiple copying of this work for scholarly purposes may be granted by me or the Dean of Graduate Studies. It is understood that copying or publication of this work for financial gain shall not be allowed without my written permission. Title of Thesis *- Pro~ertiesof Indium Arsenide Measured by Low Temperature Photoluminescence. -

INDIUM PHOSPHIDE 1. Exposure Data
pp197-226.qxd 31/05/2006 09:38 Page 197 INDIUM PHOSPHIDE 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 22398-80-7 Deleted CAS Reg. No.: 1312-40-9, 99658-38-5, 312691-22-8 Chem. Abstr. Serv. Name: Indium phosphide (InP) IUPAC Systematic Name: Indium phosphide Synonyms: Indium monophosphide 1.1.2 Molecular formula and relative molecular mass InP Relative molecular mass: 145.79 1.1.3 Chemical and physical properties of the pure substance (a) Description: Black cubic crystals (Lide, 2003) (b) Melting-point: 1062 °C (Lide, 2003) (c) Density: 4.81 g/cm3 (Lide, 2003) (d) Solubility: Slightly soluble in acids (Lide, 2003) (e) Reactivity: Can react with moisture or acids to liberate phosphine (PH3); when heated to decomposition, it may emit toxic fumes of POx (ESPI, 1994) 1.1.4 Technical products and impurities No data were available to the Working Group. 1.1.5 Analysis Occupational exposure to indium phosphide can be determined by measurement of the indium concentration in workplace air or by biological monitoring of indium. No analytical –197– pp197-226.qxd 31/05/2006 09:38 Page 198 198 IARC MONOGRAPHS VOLUME 86 methods are available for determination of indium phosphide per se. Determination of phosphorus cannot provide the required information on occupational exposure. (a) Workplace air monitoring The respirable fraction of airborne indium, collected by drawing air through a membrane filter in a stationary or personal sampler, can be determined by nondestructive, INAA. This technique has been applied to the determination of indium concentration in ambient air particulates (Kucera et al., 1999). -
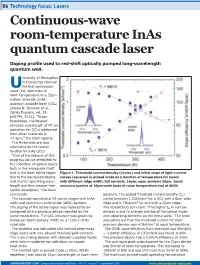
Continuous-Wave Room-Temperature Inas Quantum Cascade Laser
86 Technology focus: Lasers Continuous-wave room-temperature InAs quantum cascade laser Doping profile used to red-shift optically pumped long-wavelength quantum well. niversity of Montpellier in France has claimed Uthe first continuous- wave (cw) operation at room temperature of a 15µm indium arsenide (InAs) quantum cascade laser (QCL) [Alexei N. Baranov et al, Optics Express, vol. 24, p18799, 2016]. “To our knowledge, the longest emission wavelength of RT cw operation for QCLs fabricated from other materials is 12.4µm,” the team reports. The thresholds are also claimed to be the lowest to-date for InAs QCLs. “One of the reasons of this progress can be attributed to the reduction of optical losses both in the waveguide itself and in the laser active region Figure 1. Threshold current density (circles) and initial slope of light-current due to the decreased doping curves (squares) in pulsed mode as a function of temperature for lasers and shorter operating wave- with different ridge width: full symbols, 16µm; open symbols 20µm. Inset: length and thus weaker free emission spectra of 16µm-wide laser at room temperature and at 400K. carrier absorption,” the team comments. perature. The pulsed threshold current density (Jth) The cascade consisted of 55 active stages with InAs varied between 1.22kA/cm2 for a QCL with a 8µm-wide wells and aluminium antimonide (AlSb) barriers. ridge and 0.73kA/cm2 for one with a 20µm ridge. The doping of the active region was reduced by 6x The researchers comment: “The higher Jth in narrow compared with a previous device reported by the devices is due to a larger overlap of the optical mode same researchers. -

Gallium Arsenide
pp 163-196.qxp 31/05/2006 10:18 Page 163 GALLIUM ARSENIDE 1. Exposure Data 1.1 Chemical and physical data 1.1.1 Nomenclature Chem. Abstr. Serv. Reg. No.: 1303-00-0 Deleted CAS Reg. No.: 12254-95-4, 106495-92-5, 116443-03-9, 385800-12-4 Chem. Abstr. Serv. Name: Gallium arsenide (GaAs) IUPAC Systematic Name: Gallium arsenide Synonyms: Gallium monoarsenide 1.1.2 Molecular formula and relative molecular mass GaAs Relative molecular mass: 144.6 1.1.3 Chemical and physical properties of the pure substance (a) Description: Grey, cubic crystals (Lide, 2003) (b) Melting-point: 1238 °C (Lide, 2003) (c) Density: 5.3176 g/cm3 (Lide, 2003) (d) Solubility: Insoluble in water (Wafer Technology Ltd, 1997); slightly soluble in 0.1 M phosphate buffer at pH 7.4 (Webb et al., 1984) (e) Stability: Decomposes with evolution of arsenic vapour at temperatures above 480 °C (Wafer Technology Ltd, 1997) (f) Reactivity: Reacts with strong acid reducing agents to produce arsine gas (Wafer Technology Ltd, 1997) 1.1.4 Technical products and impurities Purity requirements for the raw materials used to produce gallium arsenide are stringent. For optoelectronic devices (light-emitting diodes (LEDs), laser diodes, photo- detectors, solar cells), the gallium and arsenic must be at least 99.9999% pure; for –163– pp 163-196.qxp 31/05/2006 10:18 Page 164 164 IARC MONOGRAPHS VOLUME 86 integrated circuits, a purity of 99.99999% is required. These purity levels are referred to by several names: 99.9999%-pure gallium is often called 6-nines, 6N or optoelectronic grade, while 99.99999%-pure gallium is called 7-nines, 7N, semi-insulating (SI) or integrated circuit (IC) grade. -

Family Type and Incidence
STRAINED INDIUM ARSENIDE/GALLIUM ARSENIDE LAYER FOR QUANTUM CASCADE LASER DESIGN USING GENETIC ALGORITHM David Mueller Dr. Gregory Triplett, Dissertation Supervisor ABSTRACT Achieving high power, continuous wave, room temperature operation of midinfrared (3-5 um) lasers is diffcult due to the effects of auger recombination in band-to-band designs. Intersubband laser designs such as quantum cascade lasers reduce the effects of recombination, increasing efficiency and have advantages in large tunability of wavelength ranges. Highly efficient quantum cascade laser designs are typically used in lasers designed for >5 um wavelength operation due to the small offset of conduction band energy in lattice matched materials. Some promising material systems have been used to achieve high-power output in the first atmospheric window (3-5 um) but still suffer from low efficiency. Larger ıconduction band offset is attainable through the use of strained materials. However, these material systems have limitations on the traditional (100) crystal orientation due to the large strain and low critical thickness. The necessity for controlled two-dimensional, optical quality layer growth limits the amount of strain incorporation due to defect formation in highly lattice mis-matched layers. The material systems used in this study are GaAs (100) and (111)B, AlGaAs, and (Ga)InAs. In the initial stage of research, I found that pseudomorphic growth of highly strained InAs layers on GaAs (111)B is possible. However, the growth window is very narrow and necessitates precise control over growth temperature and anion overpressure to achieve optical quality layers. As a result, a second stage of research explores the design space made available by this finding by using genetic algorithm based design and simulation of devices with a Schrodinger-Poisson solver. -

(12) United States Patent (10) Patent No.: US 7,166,734 B2 Shenai-Khatkhate Et Al
USOO7166734B2 (12) United States Patent (10) Patent No.: US 7,166,734 B2 Shenai-Khatkhate et al. (45) Date of Patent: Jan. 23, 2007 (54) METHOD FOR MAKING 3,808.207 A 4/1974 Shepherd et al. ........... 260/247 ORGANOMETALLC COMPOUNDS 4,364,872 A 12, 1982 Diefenbach ............. 260,448 A 4,364,873. A 12/1982 Difenbach . ... 260/448 A (75) Inventors: Deodatta Vinayak Shenai-Khatkhate, i.e. A t NG et al. O's...b wk 4 C. a. - - - RNA W. East S 4,841,082. A 6/1989 Eidt et al. ......... ... 556,129 mamchyan, Wakefield, MA (US) 4,847,399 A 7/1989 Hallock et al. ................ 556.1 5,350,869 A 9/1994 Kanjolia et al. ............... 556.1 (73) Assignee: Rohm and Haas Electronic Materials 5,473,090 A 12, 1995 Smit et al. ..................... 556.1 LLC, Marlborough, MA (US) 5,543,537 A 8/1996 Eisenberg et al. ... 556,157 - 0 5,663,390 A 9, 1997 Giolando ....................... 556.1 (*) Notice: Subject to any disclaimer, the term of this 5,756,786 A 5/1998 Power et al. .. ... 556.1 patent is extended or adjusted under 35 5,817.847. A 10, 1998 Giolando ....................... 556.1 U.S.C. 154(b) by 0 days. 6,660,874 B2 12/2003 Shenai-Khatkhate et al. .. 556/70 6,680,397 B2 1/2004 Shenai-Khatkhate et al. .. 556/1 (21) Appl. No.: 11/219,227 6,770,769 B2 8, 2004 Shenai-Khatkhate et al. .. 556/1 2004/O198042 A1 10, 2004 Shanai-Khatkhate (22) Filed: Sep. -

Phd Thesis Martin Bjergfelt
UNIVERSITY OF COPENHAGEN FACULTY OF SCIENCE Center for Quantum Devices PhD thesis Martin Bjergfelt In-situ patterned superconductor/semiconductor nanowires for quantum devices Supervisor: Professor Jesper Nygard˚ Co-supervisors: Associate Professor Thomas Sand Jespersen & Elisabetta Maria Fiordaliso This thesis has been submitted to the PhD School of The Faculty of Science, University of Copenhagen on December 20, 2019 2 Abstract The quality of interfaces and surfaces is crucial for the performance of nanoscale devices. A pertinent ex- ample is the close tie between current progress in gate-tunable and topological superconductivity using su- perconductor/semiconductor electronic devices and the hard proximity-induced superconducting gap obtained from epitaxial aluminium/indium arsenide heterostructures. Fabrication of devices requires selectively etch- ing superconductor segments from the semiconductor; this is currently only possible with Al/InAs, curbing the functional use of other superconductor/semiconductor material combinations. This thesis presents a new crystal growth platform based on three-dimensional structuring of growth substrates which is independent of the choice of semiconductor and superconductor, and enables the synthesis of semiconductor nanowires with in-situ patterned superconductor shells. This wafer-scale technique enables realisation of all the most frequently used architectures in superconducting hybrid devices. Characterisation using electron transport at cryogenic temperatures revealed an increased device yield, electrostatic stability, along with evidence of ballistic super- conductivity compared to etch-processed devices. In addition to aluminium, indium arsenide nanowires with patterned indium, vanadium, tantalum and niobium shells are presented. Structural characterisation of the hybrid nanowires by transmission electron microscopy was used to cor- relate the crystallinity of the superconductor shells to the superconducting transport properties.