Computational Modeling to Design and Analyze Synthetic Metabolic Circuits
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Rosemary)-Derived Ingredients As Used in Cosmetics
Safety Assessment of Rosmarinus Officinalis (Rosemary)-Derived Ingredients as Used in Cosmetics Status: Tentative Amended Report for Public Comment Release Date: March 28, 2014 Panel Meeting Date: June 9-10, 2014 All interested persons are provided 60 days from the above release date to comment on this safety assessment and to identify additional published data that should be included or provide unpublished data which can be made public and included. Information may be submitted without identifying the source or the trade name of the cosmetic product containing the ingredient. All unpublished data submitted to CIR will be discussed in open meetings, will be available at the CIR office for review by any interested party and may be cited in a peer-reviewed scientific journal. Please submit data, comments, or requests to the CIR Director, Dr. Lillian J. Gill. The 2014 Cosmetic Ingredient Review Expert Panel members are: Chairman, Wilma F. Bergfeld, M.D., F.A.C.P.; Donald V. Belsito, M.D.; Ronald A. Hill, Ph.D.; Curtis D. Klaassen, Ph.D.; Daniel C. Liebler, Ph.D.; James G. Marks, Jr., M.D.; Ronald C. Shank, Ph.D.; Thomas J. Slaga, Ph.D.; and Paul W. Snyder, D.V.M., Ph.D. The CIR Director is Lillian J. Gill, D.P.A. This safety assessment was prepared by Monice M. Fiume, Assistant Director/Senior Scientific Analyst. © Cosmetic Ingredient Review 1620 L Street, NW, Suite 1200♢ Washington, DC 20036 ♢ ph 202.331.0651 ♢ fax 202.331.0088 ♢ [email protected] TABLE OF CONTENTS Abstract ...................................................................................................................................................................................................................................... -

Meet Program (PDF)
SUMMER 2014NATIONALS AugustAugust 13-1713-17 UniversityUniversit ofy o Marylandf Maryland PRESENTEDPRESE N TED BY Timeline Start Times Women Event Start Times Men Wednesday, August 13, 2014 8:00 a.m. 1: mixed 1500 free (deck seeded) 8:00 a.m. Approximate finish time 1:30 p.m. Thursday, August 14, 2014 8:00 a.m. 3-4: 200 butterfly (deck seeded) 8:40 a.m. 9:15 a.m. 5-6: 50 breaststroke 9:40 a.m. 10:00 a.m. 7-8: 100 freestyle 10:40 a.m. 11:25 a.m. 9: mixed 800 freestyle (deck seeded) 11:25 a.m. Approximate finish time 3:00 p.m. Friday, August 15, 2014 8:00 a.m. 11-12: 400 IM (deck seeded) 9:10 a.m. 10:35 a.m. 30-minute warm-up in competition pool 10:35 a.m. 11:05 a.m. 13-14: 50 backstroke 11:35 a.m. 12:05 p.m. 15-16: 200 freestyle (deck seeded) 1:05 p.m. 2:10 p.m. 17-18: 100 breaststroke 2:55 p.m. 3:40 p.m. 19-20: 200 freestyle relay 4:00 p.m. Approximate finish time 4:30 p.m. Saturday, August 16, 2014 8:00 a.m. 21-22: 100 butterfly 8:30 a.m. 9:05 a.m. 23-24: 50 freestyle 9:35 a.m. 10:10 a.m. 25-26: 200 backstroke (deck seeded) 11:10 a.m. 12:10 p.m. 27: mixed 200 freestyle relay 12:10 p.m. 12:45 p.m. -

The Functional Characterization of a Site-Specific Apigenin 4
molecules Article The Functional Characterization of a Site-Specific Apigenin 40-O-methyltransferase Synthesized by the Liverwort Species Plagiochasma appendiculatum Hui Liu, Rui-Xue Xu, Shuai Gao and Ai-Xia Cheng * Key Laboratory of Chemical Biology of Natural Products, Ministry of Education, School of Pharmaceutical Sciences, Shandong University, Jinan 250012, China; [email protected] (H.L.); [email protected] (R.-X.X.); [email protected] (S.G.) * Correspondence: [email protected]; Tel.: +86-531-8838-2012; Fax: +86-531-8838-2019 Academic Editors: Qing-Wen Zhang and Chuangchuang Li Received: 5 April 2017; Accepted: 4 May 2017; Published: 7 May 2017 Abstract: Apigenin, a widely distributed flavone, exhibits excellent antioxidant, anti-inflammatory, and antitumor properties. In addition, the methylation of apigenin is generally considered to result in better absorption and greatly increased bioavailability. Here, four putative Class II methyltransferase genes were identified from the transcriptome sequences generated from the liverwort species Plagiochasma appendiculatum. Each was heterologously expressed as a His-fusion protein in Escherichia coli and their methylation activity against apigenin was tested. One of the four Class II OMT enzymes named 40-O-methyltransferase (Pa40OMT) was shown to react effectively with apigenin, catalyzing its conversion to acacetin. Besides the favorite substrate apigenin, the recombinant PaF40OMT was shown to catalyze luteolin, naringenin, kaempferol, quercetin, genistein, scutellarein, and genkwanin to the corresponding 40-methylation products. In vivo feeding experiments indicated that PaF40OMT could convert apigenin to acacetin efficiently in E. coli and approximately 88.8 µM (25.2 mg/L) of product was synthesized when 100 µM of apigenin was supplemented. -

CA Students Urge Assembly Members to Pass AB
May 26, 2021 The Honorable Members of the California State Assembly State Capitol Sacramento, CA 95814 RE: Thousands of CA Public School Students Strongly Urge Support for AB 101 Dear Members of the Assembly, We are a coalition of California high school and college students known as Teach Our History California. Made up of the youth organizations Diversify Our Narrative and GENup, we represent 10,000 youth leaders from across the State fighting for change. Our mission is to ensure that students across California high schools have meaningful opportunities to engage with the vast, diverse, and rich histories of people of color; and thus, we are in deep support of AB101 which will require high schools to provide ethnic studies starting in academic year 2025-26 and students to take at least one semester of an A-G approved ethnic studies course to graduate starting in 2029-30. Our original petition made in support of AB331, linked here, was signed by over 26,000 CA students and adult allies in support of passing Ethnic Studies. Please see appended to this letter our letter in support of AB331, which lists the names of all our original petition supporters. We know AB101 has the capacity to have an immense positive impact on student education, but also on student lives as a whole. For many students, our communities continue to be systematically excluded from narratives presented to us in our classrooms. By passing AB101, we can change the precedent of exclusion and allow millions of students to learn the histories of their peoples. -

The Phytochemistry of Cherokee Aromatic Medicinal Plants
medicines Review The Phytochemistry of Cherokee Aromatic Medicinal Plants William N. Setzer 1,2 1 Department of Chemistry, University of Alabama in Huntsville, Huntsville, AL 35899, USA; [email protected]; Tel.: +1-256-824-6519 2 Aromatic Plant Research Center, 230 N 1200 E, Suite 102, Lehi, UT 84043, USA Received: 25 October 2018; Accepted: 8 November 2018; Published: 12 November 2018 Abstract: Background: Native Americans have had a rich ethnobotanical heritage for treating diseases, ailments, and injuries. Cherokee traditional medicine has provided numerous aromatic and medicinal plants that not only were used by the Cherokee people, but were also adopted for use by European settlers in North America. Methods: The aim of this review was to examine the Cherokee ethnobotanical literature and the published phytochemical investigations on Cherokee medicinal plants and to correlate phytochemical constituents with traditional uses and biological activities. Results: Several Cherokee medicinal plants are still in use today as herbal medicines, including, for example, yarrow (Achillea millefolium), black cohosh (Cimicifuga racemosa), American ginseng (Panax quinquefolius), and blue skullcap (Scutellaria lateriflora). This review presents a summary of the traditional uses, phytochemical constituents, and biological activities of Cherokee aromatic and medicinal plants. Conclusions: The list is not complete, however, as there is still much work needed in phytochemical investigation and pharmacological evaluation of many traditional herbal medicines. Keywords: Cherokee; Native American; traditional herbal medicine; chemical constituents; pharmacology 1. Introduction Natural products have been an important source of medicinal agents throughout history and modern medicine continues to rely on traditional knowledge for treatment of human maladies [1]. Traditional medicines such as Traditional Chinese Medicine [2], Ayurvedic [3], and medicinal plants from Latin America [4] have proven to be rich resources of biologically active compounds and potential new drugs. -

CUNY Baccalaureate for Unique and Interdisciplinary Studies
Greetings from the President May 30, 2018 Dear Graduates: Congratulations! You have reached a most significant milestone in your life. Your hard work, determination, and commitment to your education have been rewarded, and you and your loved ones should take pride in your accomplishments and successes. Hunter College certainly takes pride in you. ' Your Hunter education has prepared you to meet the challenges of a world that is rapidly changing politically, socially, economically, ~ . technologically. As part of the next generation of thoughtful, responsible, and intelligent leacfets;·: you will make a real difference wherever you apply your knowledge and skills. Endless 'Opportunities await you. As you pursue your goals and move forward with your professional and personal lives., please carry with you Hunter's commitment to community, diversity, and service to others. We look forward to hearing great things about you, and we hope you will stay connected to the exciting activities and developments on campus. Please remember Hunter College and know that you will always be part of our family. Best wishes for continued success. Sincerely, ~vi Jennifer J. Raab President Order ofExercises Presiding Jennifer J. Raab, President Eija Ayravainen, Vice President for Student Affairs and Dean ofStudents Opening Ceremony Michael F. Mazzeo, Macaulay Honors College, Bachelor ofArts '18 Processional President's Party and Members of the Faculty Graduates and Candidates for Graduation National Anthem Joanna Malaszczyk, Master ofArts '18 Bagpiper Ian A. Sherman, Doctor ofNursing Practice '18 Greetings William C. Thompson, Jr., Chair, Board ofTrustees of The City University ofNew York Matthew Sapienza, Senior Vice Chancellor and ChiefFinancial Officer of The City University ofNew York ' Charge to the Graduates and Candidates for Graduation President Jennifer J. -

FEI 2016 Olympic Athlete-Horse Combination Ranking - Jumping Final : Include Events from 1.1.2015 to 06.03.2016
FEI 2016 Olympic Athlete-Horse Combination Ranking - Jumping Final : Include events from 1.1.2015 to 06.03.2016 15 best results to count over the period of 1.1.2015 to 6.3.2016 Individual athletes (VEN, COL,PER,URU, TPE) or Athletes of nations already qualified (BRA, NED, FRA, USA, GER, SWE, CAN, ARG, QAT, UKR, JPN, AUS, SUI, GBR,ESP), are not included Rank Rider NFHorse Points Olvmpic Group A 15 Bertram ALLEN IRL A MOLLY MALONE V 1187.69 27 Denis LYNCH IRL A ALL STAR 5 1092.50 33 Bertram ALLEN IRL A ROMANOV 1065.00 69 Bertram ALLEN IRL A QUIET EASY 4 855.00 87 Conor SWAIL IRL A VIVA COLOMBIA 802.50 90 Denis LYNCH IRL A ABBERVAIL VAN HET DINGESHOF 795.00 97 Dermott LENNON IRL A LOUGHVIEW LOU-LOU 780.50 122 Conor SWAIL IRL A SIMBA DE LA ROQUE 709.25 126 Cian O'CONNOR IRL A GOOD LUCK 702.50 131 Kevin BABINGTON IRL A SHORAPUR 690.50 133 Shane SWEETNAM IRL A CHAQUI Z 686.00 149 Billy TWOMEY IRL A TINKA'S SERENADE 660.00 150 Anna-Julia KONTIO FIN A PACIFIC DES ESSARTS 655.50 151 Richie MOLONEY IRL A CARRABIS Z 650.00 156 Greg Patrick BRODERICK IRL A MHS GOING GLOBAL 642.50 175 Andreas SCHOU DEN A ALLSTAR 610.00 184 Shane SWEETNAM IRL A CYKLON 1083 603.25 185 Darragh KENNY IRL A SANS SOUCIS Z 600.00 197 Niall TALBOT IRL A NICOS DE LA CENSE 590.00 222 Conor SWAIL IRL A GRAFTON 558.00 223 Conor SWAIL IRL A MARTHA LOUISE 556.75 244 Anna-Julia KONTIO FIN A FARDON 530.50 245 Conor SWAIL IRL A GRAND CRU VD VIJF EIKEN 530.00 263 Cameron HANLEY IRL A ANTELLO Z 509.00 302 Kevin BABINGTON IRL A MARK Q 477.25 307 Billy TWOMEY IRL A ARDCOLUM DUKE 472.25 -
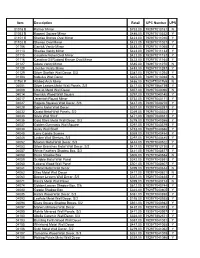
Item Description Retail UPC Number UPS 01018 B Palmer Mirror
Item Description Retail UPC Number UPS 01018 B Palmer Mirror $783.00 792977010181 N 01053 B Kagami Square Mirror $486.00 792977010532 Y 01101 B Sherise Bronze Oval Mirror $423.00 792977011010 Y 01102 B Sherise Oval Mirror $423.00 792977011027 Y 01106 Garrick Vanity Mirror $483.00 792977011065 Y 01113 Sherise Vanity Mirror $423.00 792977011133 Y 01115 Casalina Nickel Oval Mirror $423.00 792977011157 Y 01116 Casalina Oil Rubbed Bronze Oval Mirror $423.00 792977011164 Y 01127 Adara Vanity Mirror $585.00 792977011270 N 01128 Jacklyn Vanity Mirror $483.00 792977011287 Y 01129 Silver Starfish Wall Decor, S/3 $387.00 792977011294 Y 01303 Nebulus Wall Decor $405.00 792977013038 N 01760 P Ribbed Arch Mirror $486.00 792977001769 N 04001 Silver Leaves Metal Wall Panels, S/2 $417.00 792977864739 Y 04009 Ottavio Metal Wall Decor $357.00 792977040096 N 04014 Rennick Wood Wall Square $297.00 792977040140 Y 04017 Jeremiah Round Mirror $732.00 792977040171 Y 04027 Rogero Squares Wall Decor, S/6 $447.00 792977040270 Y 04028 Dorrin Metal Wall Decor $237.00 792977040287 Y 04032 Quaid Metal Wall Panels, S/2 $249.00 792977040324 Y 04033 Silvia Wall Shelf $471.00 792977040331 Y 04035 Gold Stars Metal Wall Decor, S/3 $279.00 792977040355 Y 04037 Golden Gymnasts Wall Square $297.00 792977040379 Y 04038 Auley Wall Shelf $732.00 792977040386 Y 04045 Loire Candle Sconce $369.00 792977040454 Y 04048 Lindee Wall Shelves, S/3 $297.00 792977040485 Y 04052 Maxton Metal Wall Decor, S/3 $444.00 792977040522 Y 04053 Silver Branches Metal Wall Decor, S/2 $417.00 792977812730 -

Cancer Immunology of Cutaneous Melanoma: a Systems Biology Approach
Cancer Immunology of Cutaneous Melanoma: A Systems Biology Approach Mindy Stephania De Los Ángeles Muñoz Miranda Doctoral Thesis presented to Bioinformatics Graduate Program at Institute of Mathematics and Statistics of University of São Paulo to obtain Doctor of Science degree Concentration Area: Bioinformatics Supervisor: Prof. Dr. Helder Takashi Imoto Nakaya During the project development the author received funding from CAPES São Paulo, September 2020 Mindy Stephania De Los Ángeles Muñoz Miranda Imunologia do Câncer de Melanoma Cutâneo: Uma abordagem de Biologia de Sistemas VERSÃO CORRIGIDA Esta versão de tese contém as correções e alterações sugeridas pela Comissão Julgadora durante a defesa da versão original do trabalho, realizada em 28/09/2020. Uma cópia da versão original está disponível no Instituto de Matemática e Estatística da Universidade de São Paulo. This thesis version contains the corrections and changes suggested by the Committee during the defense of the original version of the work presented on 09/28/2020. A copy of the original version is available at Institute of Mathematics and Statistics at the University of São Paulo. Comissão Julgadora: • Prof. Dr. Helder Takashi Imoto Nakaya (Orientador, Não Votante) - FCF-USP • Prof. Dr. André Fujita - IME-USP • Profa. Dra. Patricia Abrão Possik - INCA-Externo • Profa. Dra. Ana Carolina Tahira - I.Butantan-Externo i FICHA CATALOGRÁFICA Muñoz Miranda, Mindy StephaniaFicha de Catalográfica los Ángeles M967 Cancer immunology of cutaneous melanoma: a systems biology approach = Imuno- logia do câncer de melanoma cutâneo: uma abordagem de biologia de sistemas / Mindy Stephania de los Ángeles Muñoz Miranda, [orientador] Helder Takashi Imoto Nakaya. São Paulo : 2020. 58 p. -

Bio::Graphics HOWTO Lincoln Stein Cold Spring Harbor Laboratory1 [email protected]
Bio::Graphics HOWTO Lincoln Stein Cold Spring Harbor Laboratory1 [email protected] This HOWTO describes how to render sequence data graphically in a horizontal map. It applies to a variety of situations ranging from rendering the feature table of a GenBank entry, to graphing the positions and scores of a BLAST search, to rendering a clone map. It describes the programmatic interface to the Bio::Graphics module, and discusses how to create dynamic web pages using Bio::DB::GFF and the gbrowse package. Table of Contents Introduction ...........................................................................................................................3 Preliminaries..........................................................................................................................3 Getting Started ......................................................................................................................3 Adding a Scale to the Image ...............................................................................................5 Improving the Image............................................................................................................7 Parsing Real BLAST Output...............................................................................................8 Rendering Features from a GenBank or EMBL File ....................................................12 A Better Version of the Feature Renderer ......................................................................14 Summary...............................................................................................................................18 -

UCLA UCLA Electronic Theses and Dissertations
UCLA UCLA Electronic Theses and Dissertations Title Bipartite Network Community Detection: Development and Survey of Algorithmic and Stochastic Block Model Based Methods Permalink https://escholarship.org/uc/item/0tr9j01r Author Sun, Yidan Publication Date 2021 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA Los Angeles Bipartite Network Community Detection: Development and Survey of Algorithmic and Stochastic Block Model Based Methods A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Statistics by Yidan Sun 2021 © Copyright by Yidan Sun 2021 ABSTRACT OF THE DISSERTATION Bipartite Network Community Detection: Development and Survey of Algorithmic and Stochastic Block Model Based Methods by Yidan Sun Doctor of Philosophy in Statistics University of California, Los Angeles, 2021 Professor Jingyi Li, Chair In a bipartite network, nodes are divided into two types, and edges are only allowed to connect nodes of different types. Bipartite network clustering problems aim to identify node groups with more edges between themselves and fewer edges to the rest of the network. The approaches for community detection in the bipartite network can roughly be classified into algorithmic and model-based methods. The algorithmic methods solve the problem either by greedy searches in a heuristic way or optimizing based on some criteria over all possible partitions. The model-based methods fit a generative model to the observed data and study the model in a statistically principled way. In this dissertation, we mainly focus on bipartite clustering under two scenarios: incorporation of node covariates and detection of mixed membership communities. -

Stein Gives Bioinformatics Ten Years to Live
O'Reilly Network: Stein Gives Bioinformatics Ten Years to Live http://www.oreillynet.com/lpt/a/3205 Published on O'Reilly Network (http://www.oreillynet.com/) http://www.oreillynet.com/pub/a/network/biocon2003/stein.html See this if you're having trouble printing code examples Stein Gives Bioinformatics Ten Years to Live by Daniel H. Steinberg 02/05/2003 Lincoln Stein's keynote at the O'Reilly Bioinformatics Technology Conference was provocatively titled "Bioinformatics: Gone in 2012." Despite the title, Stein is optimistic about the future for people doing bioinformatics. But he explained that "the field of bioinformatics will be gone by 2012. The field will be doing the same thing but it won't be considered a field." His address looked at what bioinformatics is and what its future is likely to be in the context of other scientific disciplines. He also looked at career prospects for people doing bioinformatics and provided advice for those looking to enter the field. What Is Bioinformatics: Take One Stein, of the Cold Spring Harbor Laboratory, began his keynote by examining what is meant by bioinformatics. In the past such a talk would begin with a definition displayed from an authoritative dictionary. The modern approach is to appeal to an FAQ from an authoritative web site. Take a look at the FAQ at bioinformatics.org and you'll find several definitions. Stein summarized Fedj Tekaia of the Institut Pasteur--that bioinformatics is DNA and protein analysis. Stein also summarized Richard Durbin of the Sanger Institute--that bioinformatics is managing data sets. Stein's first pass at a definition of bioinformatics is that it is "Biologists using computers or the other way around." He followed by observing that whatever it is, it's growing.