Three-Dimensional Docking of Alcohols to Ketones: an Experimental Benchmark Based on Cite This: Phys
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Acetophenone Sigma Aldrich.Pdf
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 4.0 Revision Date 02/27/2010 Print Date 07/08/2011 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Acetophenone Product Number : A10701 Brand : Sigma-Aldrich Company : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # : (314) 776-6555 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Combustible Liquid, Harmful by ingestion., Irritant GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H227 Combustible liquid H302 Harmful if swallowed. H318 Causes serious eye damage. Precautionary statement(s) P210 Keep away from heat/sparks/open flames/hot surfaces. - No smoking. P264 Wash skin thoroughly after handling. P270 Do not eat, drink or smoke when using this product. P280 Wear protective gloves/protective clothing/eye protection/face protection. P301 + P312 IF SWALLOWED: Call a POISON CENTER or doctor/physician if you feel unwell. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P310 Immediately call a POISON CENTER or doctor/physician. P330 Rinse mouth. P370 + P378 In case of fire: Use dry sand, dry chemical or alcohol-resistant foam for extinction. P403 + P235 Store in a well-ventilated place. Keep cool. P501 Dispose of contents/container to an approved waste disposal plant. HMIS Classification Health hazard: 2 Flammability: 2 Physical hazards: 0 NFPA Rating Health hazard: 2 Fire: 2 Reactivity Hazard: 0 Potential Health Effects Sigma-Aldrich - A10701 Page 1 of 6 Inhalation May be harmful if inhaled. -

ACETOPHENONE Method No. PV2003 Target Concentration
ACETOPHENONE Method no. PV2003 Target concentration: 100 ppm (491 mg/m3) Procedure: Samples are collected by drawing a known volume of air through a Tenax GC tube. Samples are desorbed with a solvent mixture of (5:95) Isopropanol:Carbon Disulfide (IPA):(CS2) and analyzed by gas chromatography with a flame ionization detector. Recommended air volume 120 minutes at 0.1 L/min (12 L) and sampling rate: Status of Method: Partially Validated. This method has been only partially evaluated and is presented for information and trial use. July 1982 Wayne Potter Service Branch 1 OSHA Salt Lake Technical Center Salt Lake City UT-84115 1 of 8 1 General Discussion 1.1 Background 1.1.1 History of procedure Recently, the OSHA Analytical Laboratory received a set of field samples that required analysis for acetophenone. The air samples had been collected on charcoal tubes and isopropanol impingers. Desorption studies were done on charcoal tubes using acetone, carbon disulfide, 5:95 isopropanol:carbon disulfide, and methylene chloride as desorbing solvents. The best results were obtained with 5:95 isopropanol:carbon disulfide, but the recovery was only 68%, which indicated that charcoal tubes should not be recommended for the collection of acetophenone air samples. An evaluation of isopropanol impingers was not performed because it was preferable to find an adsorbent procedure for sample collection. NIOSH method 291 for α -chloroacetophenone uses Tenax GC tubes. The Tenax GC tubes were tried for acetophenone, and appeared to be a suitable method of collection. 1.1.2 Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy.) Acetophenone is a narcotic in high concentrations (Ref. -
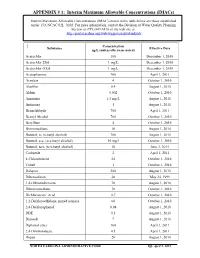
APPENDIX # 1: Interim Maximum Allowable Concentrations (Imacs)
APPENDIX # 1: Interim Maximum Allowable Concentrations (IMACs) Interim Maximum Allowable Concentrations (IMAC) shown in the table below are those established under 15A NCAC 02L .0202. For more information, contact the Division of Water Quality Planning Section at (919) 807-6416 or the web site at: http://portal.ncdenr.org/web/wq/ps/csu/gwstandards Concentration Substance Effective Date ug/L (unless otherwise noted) Acetochlor 100 December 1, 2010 Acetochlor ESA 1 mg/L December 1, 2010 Acetochlor OXA 1 mg/L December 1, 2010 Acetophenone 700 April 1, 2011 Acrolein 4 October 1, 2010 Alachlor 0.4 August 1, 2010 Aldrin 0.002 October 1, 2010 Ammonia 1.5 mg/L August 1, 2010 Antimony 1 August 1, 2010 Benzaldehyde 700 April 1, 2011 Benzyl Alcohol 700 October 1, 2010 Beryllium 4 October 1, 2010 Bromomethane 10 August 1, 2010 Butanol, n- (n-butyl alcohol) 700 August 1, 2010 Butanol, sec- (sec-butyl alcohol) 10 mg/l October 1, 2010 Butanol, tert- (tert-butyl alcohol) 10 June 1, 2011 Carbazole 2 April 1, 2011 4-Chlorotoluene 24 October 1, 2010 Cobalt 1 October 1, 2010 Dalapon 200 August 1, 2010 Dibenzofuran 28 May 24, 1999 1,4-Dibromobenzene 70 August 1, 2010 Dibromomethane 70 October 1, 2010 Dichloroacetic Acid 0.7 October 1, 2010 1,2-Dichloroethylene, mixed isomers 60 October 1, 2010 2,4-Dichlorophenol 0.98 August 1, 2010 DDE 0.1 August 1, 2010 Dinoseb 7 August 1, 2010 Diphenyl ether 100 April 1, 2011 2,4-Dinitrotoluene 0.1 April 1, 2011 Diquat 20 August 1, 2010 NORTH CAROLINA ADMINISTRATIVE CODE Eff. -

DESIGN, SYNTHESIS, CHARACTERIZATION of SOME NEW SUBSTITUTED CHALCONES and STUDIES THEIR ANTIMICROBIAL ACTIVITIES Zakaria H
[Aiube* et al., 5(6): June, 2016] ISSN: 2277-9655 IC™ Value: 3.00 Impact Factor: 4.116 IJESRT INTERNATIONAL JOURNAL OF ENGINEERING SCIENCES & RESEARCH TECHNOLOGY DESIGN, SYNTHESIS, CHARACTERIZATION OF SOME NEW SUBSTITUTED CHALCONES AND STUDIES THEIR ANTIMICROBIAL ACTIVITIES Zakaria H. Aiube*, Ali H. Samir, Israa SH. A- R. Al- Kadi * Department of Chemistry, college of Education for pure Science-Ibn, Al- Haitham, University, Baghdad, Iraq DOI: 10.5281/zenodo.55982 ABSTRACT Eight designed chalcones, named [1(p-benzenesulphonamidophenyl)-3-p-chloro-2-propene-1-one][2], [1(p- benzenesulphonamidophenyl)-3-p-nitro-2-propene-1-one][3], [1(4-Ureido)phenyl-3-p-chlorophenyl-2-propene- 1-one][5], [1(4-Ureido)phenyl-3-p-nitroophenyl-2-propene-1-one][6], [1(4(p-N-methylaminophenyl)azophenyl- 3-p-chlorophenyl-2-propene-1-one][8], [1(4(p-N-methylaminophenyl)azophenyl-3-p-nitroophenyl-2-propene-1- one][9], [1(p-aminophenyl)-3-p-chlorophenyl-2-propene-1-one][10] and [1(p-aminophenyl)-3-p-nitrophenyl-2- propene-1-one][11], were synthesised by condensation of synthesised p-acetylphenylbenzene- sulphonamide, p- acetylphenylurea and p-acetyl-p'-(N-methylamino)azobenzene, with p-chlorobenzaldehyde and p nitrobenzaldehyde in basic media respectively. All synthesised compounds are characterized by its melting points, FTIR, 1H NMR, 13C NMR and Mass spectral analysis. All synthesised compounds are examined their antimicrobial activities against Gram-Ve bactria (Serratia marcescens, Pseudmonas aeroginosa) and Gram+Ve bacterial (Stphylococcus aureus, Streptococcus pyogenes), and Candida albicans fungi. Result showed good to moderate inhibition effect against some bacteria and fungi, in comparison with some pharmaceutical antibiotic and antifungal treatments like Cephalexin, Amoxicillin, Tetracycline, Lincomycine, Nystatine and Fluconazole respectively. -

Acetophenone
Acetophenone 98-86-2 Hazard Summary Acetophenone is used for fragrance in soaps and perfumes, as a flavoring agent in foods, and as a solvent for plastics and resins. Acute (short-term) exposure to acetophenone vapor may produce skin irritation and transient corneal injury in humans. No information is available on the chronic (long-term), reproductive, developmental, or carcinogenic effects of acetophenone in humans. EPA has classified acetophenone as a Group D, not classifiable as to human carcinogenicity. Please Note: The main sources of information for this fact sheet are EPA's Integrated Risk Information System (IRIS) (5), which contains information on oral chronic toxicity of acetophenone and the RfD, and EPA's Health and Environmental Effects Document for Acetophenone (2). Other secondary sources include the Hazardous Substances Data Bank (HSDB) (3), a database of summaries of peer-reviewed literature, and the Registry of Toxic Effects of Chemical Substances (RTECS) (4), a database of toxic effects that are not peer reviewed. Uses Acetophenone is used in perfumery as a fragrance ingredient in soaps, detergents, creams, lotions, and perfumes; as a flavoring agent in foods, nonalcoholic beverages, and tobacco; as a specialty solvent for plastics and resins; as a catalyst for the polymerization of olefins; and in organic syntheses as a photosensitizer. (2,3,6) Sources and Potential Exposure Occupational exposure to acetophenone may occur during its manufacture and use. (1) Acetophenone has been detected in ambient air and drinking water; exposure of the general public may occur through the inhalation of contaminated air or the consumption of contaminated water. (2) Assessing Personal Exposure Hippuric acid may be monitored in the urine to determine whether or not exposure to acetophenone has occurred. -

United States Patent Office Patented Aug
3,461,164 United States Patent Office Patented Aug. 12, 1969 2 3,461,64 propyl-(1), 1-phenoxypropyl-(2), o-, m- or p-hydroxy HYDROXY-2AMNO-1-PHENYLALKANES benzyl, o-, m- or p-methoxybenzyl, o-, m- or p-ethoxy AND THE SALTS THEREOF benzyl, 3,4-dihydroxybenzyl, 3,4-dimethoxybenzyi, 3,4,5- Karl Schulte and Wolfgang Fruhstorfer, Darmstadt, Hein trihydroxybenzyl, 3,4,5-trimethoxybenzyl or 3.4-methyl rich Muller, Pfungstadt, Hans Friebel, Darmstadt-Eber endioxybenzyl. stadt, Hans Joachim Enenkel, Darmstadt, and Josef Particularly preferred compounds are: Gilissen, Escholbrucken, Germany, assignors to E. Merck Aktiengesellschaft, Darmstadt, Germany N-1-(3'-tert, butyl-4'-hydroxy-5'-methyl-phenyl)- No Drawing. Filed Apr. 27, 1964, Ser. No. 364,355 1-hydroxy-butyl-2-N-isopropylamine Claims priority, application Germany, Apr. 25, 1963, N-(1-(3'-tert.butyl-4'-hydroxy-5'-methyl-phenyl)- M 56,597; May 25, 1963, M 56,960 O 1-hydroxy-propyl-2)-N-methylamine Int, C. CO7 91/18 N-1-(3'-tert.butyl-4'-hydroxy-5'-methyl-phenyl)- U.S. C. 260-570.6 22 Claims 1-hydroxy-propyl-2-N-ethylamine N-(1-(3'-tert.butyl-4-hydroxy-5'-methyl-phenyl)- This invention relates to substituted 1-hydroxy-2-amino 1-hydroxy-propyl-2-N-isopropylamine 1-phenyl-alkanes. 5 N-1-(3'-tert.butyl-4'-hydroxy-5'-methyl-phenyl)- It is a principal object of this invention to provide novel 1-hydroxy-propyl-2-N-(4-phenyl-butyl-2)-amine and unobvious chemical compounds. N-E1-(3'-tert.butyl-4'-hydroxy-5'-methyl-phenyl)- Another object is to provide processes for the produc 1-hydroxy-propyl-2-N-(1-phenoxy-propyl-2)-amine tion of these compounds. -

Purification and Characterization of an NADH-Dependent Alcohol
Biosci. Biotechnol. Biochem., 75 (6), 1055–1060, 2011 Purification and Characterization of an NADH-Dependent Alcohol Dehydrogenase from Candida maris for the Synthesis of Optically Active 1-(Pyridyl)ethanol Derivatives y Shigeru KAWANO, Miho YANO, Junzo HASEGAWA, and Yoshihiko YASOHARA Frontier Biochemical and Medicinal Research Laboratories, Kaneka Corporation, 1-8 Miyamae-machi, Takasago-cho, Takasago, Hyogo 676-8688, Japan Received July 23, 2010; Accepted March 28, 2011; Online Publication, June 13, 2011 [doi:10.1271/bbb.100528] A novel (R)-specific alcohol dehydrogenase (AFPDH) tor.6) We found that Candida maris IFO10003 has a (R)- produced by Candida maris IFO10003 was purified to specific reducing enzyme that reduces acetylpyridine homogeneity by ammonium sulfate fractionation, derivatives to (R)-1-(pyridyl)ethanol derivatives with DEAE-Toyopearl, and Phenyl-Toyopearl, and charac- more than 99% e.e.7) Here we describe the purification terized. The relative molecular mass of the native and characterization of that (R)-specific enzyme. enzyme was found to be 59,900 by gel filtration, and that of the subunit was estimated to be 28,900 on SDS- Materials and Methods polyacrylamide gel electrophoresis. These results sug- gest that the enzyme is a homodimer. It required NADH Chemicals. 5-Acetylfuro[2,3-c]pyridine (AFP) and 5-acetyl-7- as a cofactor and reduced various kinds of carbonyl chlorofuro[2,3-c]pyridine were prepared following previous reports.8,9) compounds, including ketones and aldehydes. AFPDH Racemic alcohols were prepared by NaBH4 reduction. Glucose dehydrogenase was purchased from Amano Enzyme (Aichi, Japan). reduced acetylpyridine derivatives, -keto esters, and All the other chemicals used in this study were of analytical grade and some ketone compounds with high enantioselectivity. -

Material Safety Data Sheet Acetophenone MSDS
He a lt h 1 2 Fir e 2 1 0 Re a c t iv it y 0 P e r s o n a l P r o t e c t io n J Material Safety Data Sheet Acetophenone MSDS Section 1: Chemical Product and Company Identification Product Name: Acetophenone Contact Information: Catalog Codes: SLA2425 Sciencelab.com, Inc. 14025 Smith Rd. CAS#: 98-86-2 Houston, Texas 77396 RTECS: AM5250000 US Sales: 1-800-901-7247 International Sales: 1-281-441-4400 TSCA: TSCA 8(b) inventory: Acetophenone Order Online: ScienceLab.com CI#: Not applicable. CHEMTREC (24HR Emergency Telephone), call: Synonym: Ketone methyl phenyl 1-800-424-9300 Chemical Name: 1-Phenyl-ethanone International CHEMTREC, call: 1-703-527-3887 Chemical Formula: C6H5COCH4 For non-emergency assistance, call: 1-281-441-4400 Section 2: Composition and Information on Ingredients Composition: Name CAS # % by Weight Acetophenone 98-86-2 100 Toxicological Data on Ingredients: Acetophenone: ORAL (LD50): Acute: 815 mg/kg [Rat.]. 740 mg/kg [Mouse]. DERMAL (LD50): Acute: 15900 mg/kg [Rabbit]. Section 3: Hazards Identification Potential Acute Health Effects: Very hazardous in case of eye contact (irritant). Hazardous in case of skin contact (irritant). Slightly hazardous in case of ingestion, of inhalation. Inflammation of the eye is characterized by redness, watering, and itching. Potential Chronic Health Effects: Very hazardous in case of eye contact (irritant). Hazardous in case of skin contact (irritant). Slightly hazardous in case of ingestion, of inhalation. CARCINOGENIC EFFECTS: Not available. MUTAGENIC EFFECTS: Not available. TERATOGENIC EFFECTS: Not available. DEVELOPMENTAL TOXICITY: Not available. -

Identification of an Unknown – Alcohols, Aldehydes, and Ketones
Identification of an Unknown: Alcohols, Aldehydes, and Ketones How does one determine the identity and structure of an unknown compound? This is not a trivial task. Modern x-ray and spectroscopic techniques have made the job much easier, but for some very complex molecules, identification and structure determination remains a challenge. In addition to spectroscopic information and information obtained from other instrumental methods, chemical reactions can provide useful structural information, and physical properties can contribute significantly to confirming the identity of a compound. In this experiment, you will be asked to identify an unknown liquid, which will be either an alcohol, aldehyde, or ketone. Identification will be accomplished by carrying out chemical tests, called classification tests, preparing a solid derivative of the unknown and determining its melting point (MP), making careful observations, and analyzing the NMR spectrum of the unknown. the carbonyl group O O R OH R H R R alcohol aldehyde ketone A list of alcohols, aldehydes, and ketones, along with the MP of a solid derivative of each compound, is posted on the website. The unknown will be one of these listed compounds. If one can determine to which functional group class (alcohol, aldehyde, or ketone) the unknown belongs, two of the three lists need not be considered and the task will be greatly simplified. To accomplish this, classification tests will be carried out. First, consider some general ways in which alcohols, aldehydes, and ketones react. 1 CLASSIFICATION TESTS, which are simple chemical reactions that produce color changes or form precipitates, can be used to differentiate alcohols, aldehydes, and ketones, and also to provide further structural information. -

The Oxidation of Acetophenone to Produce Benzoic Acid AEM – Last Update July 2017
The Oxidation of Acetophenone to Produce Benzoic Acid AEM – last update July 2017 The oxidation of acetophenone to form benzoic acid is the first step in the three labs that together make up our multistep synthesis project. EACH STEP OF THIS PROJECT WILL BE WRITTEN UP AS SEPARATE LABS, but you will use the products from the early steps as reagents for the subsequent steps. Each product must be carefully characterized (by mp, IR, and/or NMR) before using it in the next step. This multi-step synthesis is drawn from a Journal of Chemical Education Article called “And the Winner is… A Multi-Step Synthesis for the Introductory Organic Course”. Its reference is S. S. Stradling; C. L. Gage J. Chem. Ed. 1985, 62(12), 1116-1117. We have chosen only one of the three possible synthetic routes described in this paper for preparing a single substance, methyl m- nitrobenzoate. Following the oxidation, we will perform a nitration of the aromatic ring, then finally an esterification of the carboxylic acid. Abbreviated versions of the three reaction steps are given here, with more complete versions of each of the overall reactions given with each lab prompt separately. All Three Steps of the Multi-Step Reaction Sequence: O COOH COOCH3 [O] HNO3 CH3OH Ph C CH3 PhCOOH H2SO4 H2SO4 NO2 NO2 Step 1: the Oxidation of Acetophenone to Produce Benzoic Acid Overall Reaction: O NaOCl At RT, Ph C CH3 PhCOO + CHCl3 (g) PhCOOH NaOH HCl HEAT CAUTION! You are using strong base, strong acid, and bleach in this reaction. Some of these caustic materials will be heated, which their danger. -

A Study of Chloretone and Related Compounds Earle Alexander Tompkins University of Massachusetts Amherst
University of Massachusetts Amherst ScholarWorks@UMass Amherst Masters Theses 1911 - February 2014 1933 A study of chloretone and related compounds Earle Alexander Tompkins University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/theses Tompkins, Earle Alexander, "A study of chloretone and related compounds" (1933). Masters Theses 1911 - February 2014. 2041. Retrieved from https://scholarworks.umass.edu/theses/2041 This thesis is brought to you for free and open access by ScholarWorks@UMass Amherst. It has been accepted for inclusion in Masters Theses 1911 - February 2014 by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. MASSACHUSETTS STATE COLLEGE at c 01 to DATE DUE UNIVERSITY OF MASSACHUSETTS T TRB A R V PHYS SC! A STUDY cf rm.rwmr't xm WSLXTSB Ct«i'< VBUQ Sarle Mexander Tompkins Theeie submitted for the decree of iter of nelonce MA33/.CRtrSSTTS 3TATS COLL?*» June* 19SS. • Page Introduction • ............................ 1. Theoretic il "Discuasicn and Sevier of Literature 3. Experimental Tart 9. I* "reparation of Ci loretone. • • ..10. II. Condensation of 3thyl Hethyl Tetone and Chloro form. 12 . III. Condensation of Diethyl Ketone and Chlorofom...« 16. IV. Condensation of U-I!e>:yl Jfethyl Ketone and Chloroform.. 18. V. Condensation of .'cetophenone and Chloroform. ••••• 19. VI. Condensation of Cyclohexanone and Chloroform. ... First Condensation £3* Jecond " ................. £7. Identification of Product 29. Molecular .'eight 30. Chlorine Determination 31. VII. Condensation of Canphor and Chloroform 35. 7311, Condensation of Acetone and Chloroform Using lodauide as Catalyst... 35. Conclusions. 37. Bibliography 35. Acknowledgement 40 ITTTRCDTTTirH In 1P8 , Yillgerodt (16) prepared acetone chloroform by condensing acetone with chloroform by means of pulverized potassium hydroxide.