Alternatives for the Production of Propene Oxide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The PO Production Chain and Possibilities for Energy Saving
The PO production chain and possibilities for energy saving Public Summary Report Delft, January 2012 Author(s): Ewout Dönszelmann Ab de Buck Harry Croezen Lonneke Wielders Publication Data Bibliographical data: Ewout Dönszelmann, Ab de Buck, Harry Croezen, Lonneke Wielders The PO production chain and possibilities for energy saving Public Summary Delft, CE Delft, January 2012 Energy saving / Chain Management / Products / Chemical industry / Propylene Oxide / Styrene / Iso-butylene FT: Tert-butyl alcohol (TBA) Publication code: 12.2232.12 CE publications are available from www.cedelft.eu. Commissioned by: Agentschap NL Further information on this study can be obtained from the contact person, Ewout Dönszelmann. © copyright, CE Delft, Delft CE Delft Committed to the Environment CE Delft is an independent research and consultancy organisation specialised in developing structural and innovative solutions to environmental problems. CE Delft’s solutions are characterised in being politically feasible, technologically sound, economically prudent and socially equitable. 2 January 2012 2.232.3 – The PO production chain and possibilities for energy saving Contents 1 Introduction 5 1.1 General background 5 1.2 The project 5 1.3 Approach 6 1.4 Reading guide 6 2 The production chains 7 2.1 The two processes 7 2.2 Expanded polystyrene 9 2.3 Polyols 11 2.4 TBA/iso-butylene applications 13 3 Conclusions, recommendations 17 3.1 Conclusions 17 3.2 Recommendations 17 4 Background of the Long-term Agreement Energy Efficiency ETS enterprises (LEE) 19 References 21 3 January 2012 2.232.3 – The PO production chain and possibilities for energy saving 4 January 2012 2.232.3 – The PO production chain and possibilities for energy saving 1 Introduction 1.1 General background The Dutch Industry and the Dutch government have made long term agreements on energy efficiency (LEE) for companies that are under the European Trading Scheme (ETS). -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Acetophenone Sigma Aldrich.Pdf
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 4.0 Revision Date 02/27/2010 Print Date 07/08/2011 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Acetophenone Product Number : A10701 Brand : Sigma-Aldrich Company : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # : (314) 776-6555 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Combustible Liquid, Harmful by ingestion., Irritant GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H227 Combustible liquid H302 Harmful if swallowed. H318 Causes serious eye damage. Precautionary statement(s) P210 Keep away from heat/sparks/open flames/hot surfaces. - No smoking. P264 Wash skin thoroughly after handling. P270 Do not eat, drink or smoke when using this product. P280 Wear protective gloves/protective clothing/eye protection/face protection. P301 + P312 IF SWALLOWED: Call a POISON CENTER or doctor/physician if you feel unwell. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P310 Immediately call a POISON CENTER or doctor/physician. P330 Rinse mouth. P370 + P378 In case of fire: Use dry sand, dry chemical or alcohol-resistant foam for extinction. P403 + P235 Store in a well-ventilated place. Keep cool. P501 Dispose of contents/container to an approved waste disposal plant. HMIS Classification Health hazard: 2 Flammability: 2 Physical hazards: 0 NFPA Rating Health hazard: 2 Fire: 2 Reactivity Hazard: 0 Potential Health Effects Sigma-Aldrich - A10701 Page 1 of 6 Inhalation May be harmful if inhaled. -

Removal of NO and SO2 by Pulsed Corona Discharge Process
Korean J. Chem. Eng., 18(3), 308-316 (2001) Removal of NO and SO2 by Pulsed Corona Discharge Process Young Sun Mok†, Ho Won Lee, Young Jin Hyun, Sung Won Ham*, Jae Hak Kim** and In-Sik Nam** Department of Chemical Engineering, Cheju National University, Ara, Cheju 690-756, Korea *Department of Chemical Engineering, Kyungil University, Hayang, Kyungbuk 712-701, Korea **Department of Chemical Engineering, Pohang University of Science & Technology, Pohang, Kyungbuk 790-784, Korea (Received 6 September 2000 • accepted 9 March 2001) − Abstract Overall examination was made on the removal of NO and SO2 by pulsed corona discharge process. The mechanism for the removal of NO was found to largely depend on the gas composition. In the absence of oxygen, most of the NO removed was reduced to N2; on the other hand, oxidation of NO to NO2 was dominant in the presence of oxygen even when the content was low. Water vapor was an important ingredient for the oxidation of NO2 to nitric acid rather than that of NO to NO2. The removal of NO only slightly increased with the concentration of ammonia while the effect of ammonia on the removal of SO2 was very significant. The energy density (power delivered/feed gas flow rate) can be a measure for the degree of removal of NO. Regardless of the applied voltage and the flow rate of the feed gas stream, the amount of NO removed was identical at the same energy density. The production of N2O increased with the pulse repetition rate, and the presence of NH3 and SO2 enhanced it. -

INF.20 Economic Commission for Europe Inland Transport Committee Definition Of
INF.20 Economic Commission for Europe Inland Transport Committee Working Party on the Transport of Dangerous Goods 6 March 2012 Joint Meeting of the RID Committee of Experts and the Working Party on the Transport of Dangerous Goods Bern, 19–23 March 2012 Item 5(a) of the provisional agenda Proposals for amendments to RID/ADR/ADN: pending issues Definition of LPG Transmitted by the Government of Switzerland We have observed contradictions in the definition of LPG adopted by the Joint Meeting at its Autumn 2010 session (ECE/TRANS/WP.15/AC.1/120, Annex II) as well as in the German translation we have discussed in Erfurt during the Conference of translation and edition of the German ADR. The actual definitions in each language are the following: ""Gaz de pétrole liquéfié (GPL)", un gaz liquéfié à faible pression contenant un ou plusieurs hydrocarbures légers qui sont affectés aux numéros ONU 1011, 1075, 1965, 1969 ou 1978 seulement et qui est principalement constitué de propane, de propène, de butane, des isomères du butane, de butène avec des traces d’autres gaz d’hydrocarbures. “‘Liquefied Petroleum Gas (LPG)’ means a low pressure liquefied gas composed of one or more light hydrocarbons which are assigned to UN Nos. 1011, 1075, 1965, 1969 or 1978 only and which consists mainly of propane, propene, butane, butane isomers, butene with traces of other hydrocarbon gases. „Flüssiggas (LPG)*: Unter geringem Druck verflüssigtes Gas, das aus einem oder mehreren leichten Kohlenwasserstoffen besteht, die nur der UN-Nummer 1011, 1075, 1965, 1969 oder 1978 zugeordnet sind und das hauptsächlich aus Propan, Propen, Butan, Butan-Isomeren und Buten mit Spuren anderer Kohlenwasserstoffgase besteht.“ One important point is different between French from one side and English and German from the other side, that is that in French the repetition of the “de” in the enumeration of possible gases brings to the conclusion that the main constituent of the LPG is one of the listed gases. -

ACETOPHENONE Method No. PV2003 Target Concentration
ACETOPHENONE Method no. PV2003 Target concentration: 100 ppm (491 mg/m3) Procedure: Samples are collected by drawing a known volume of air through a Tenax GC tube. Samples are desorbed with a solvent mixture of (5:95) Isopropanol:Carbon Disulfide (IPA):(CS2) and analyzed by gas chromatography with a flame ionization detector. Recommended air volume 120 minutes at 0.1 L/min (12 L) and sampling rate: Status of Method: Partially Validated. This method has been only partially evaluated and is presented for information and trial use. July 1982 Wayne Potter Service Branch 1 OSHA Salt Lake Technical Center Salt Lake City UT-84115 1 of 8 1 General Discussion 1.1 Background 1.1.1 History of procedure Recently, the OSHA Analytical Laboratory received a set of field samples that required analysis for acetophenone. The air samples had been collected on charcoal tubes and isopropanol impingers. Desorption studies were done on charcoal tubes using acetone, carbon disulfide, 5:95 isopropanol:carbon disulfide, and methylene chloride as desorbing solvents. The best results were obtained with 5:95 isopropanol:carbon disulfide, but the recovery was only 68%, which indicated that charcoal tubes should not be recommended for the collection of acetophenone air samples. An evaluation of isopropanol impingers was not performed because it was preferable to find an adsorbent procedure for sample collection. NIOSH method 291 for α -chloroacetophenone uses Tenax GC tubes. The Tenax GC tubes were tried for acetophenone, and appeared to be a suitable method of collection. 1.1.2 Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy.) Acetophenone is a narcotic in high concentrations (Ref. -
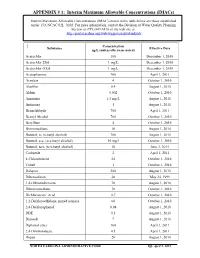
APPENDIX # 1: Interim Maximum Allowable Concentrations (Imacs)
APPENDIX # 1: Interim Maximum Allowable Concentrations (IMACs) Interim Maximum Allowable Concentrations (IMAC) shown in the table below are those established under 15A NCAC 02L .0202. For more information, contact the Division of Water Quality Planning Section at (919) 807-6416 or the web site at: http://portal.ncdenr.org/web/wq/ps/csu/gwstandards Concentration Substance Effective Date ug/L (unless otherwise noted) Acetochlor 100 December 1, 2010 Acetochlor ESA 1 mg/L December 1, 2010 Acetochlor OXA 1 mg/L December 1, 2010 Acetophenone 700 April 1, 2011 Acrolein 4 October 1, 2010 Alachlor 0.4 August 1, 2010 Aldrin 0.002 October 1, 2010 Ammonia 1.5 mg/L August 1, 2010 Antimony 1 August 1, 2010 Benzaldehyde 700 April 1, 2011 Benzyl Alcohol 700 October 1, 2010 Beryllium 4 October 1, 2010 Bromomethane 10 August 1, 2010 Butanol, n- (n-butyl alcohol) 700 August 1, 2010 Butanol, sec- (sec-butyl alcohol) 10 mg/l October 1, 2010 Butanol, tert- (tert-butyl alcohol) 10 June 1, 2011 Carbazole 2 April 1, 2011 4-Chlorotoluene 24 October 1, 2010 Cobalt 1 October 1, 2010 Dalapon 200 August 1, 2010 Dibenzofuran 28 May 24, 1999 1,4-Dibromobenzene 70 August 1, 2010 Dibromomethane 70 October 1, 2010 Dichloroacetic Acid 0.7 October 1, 2010 1,2-Dichloroethylene, mixed isomers 60 October 1, 2010 2,4-Dichlorophenol 0.98 August 1, 2010 DDE 0.1 August 1, 2010 Dinoseb 7 August 1, 2010 Diphenyl ether 100 April 1, 2011 2,4-Dinitrotoluene 0.1 April 1, 2011 Diquat 20 August 1, 2010 NORTH CAROLINA ADMINISTRATIVE CODE Eff. -

Plasma-Pd/Γ-Al203 Catalytic System for Methane, Toluene and Propene Oxidation: Effect of Temperature and Plasma Input Power
22nd International Symposium on Plasma Chemistry July 5-10, 2015; Antwerp, Belgium Plasma-Pd/γ-Al203 catalytic system for methane, toluene and propene oxidation: effect of temperature and plasma input power T. Pham Huu1, 2, P. Da Costa3, S. Loganathan1 and A. Khacef1 1 GREMI-UMR 6744, CNRS-Université d'Orléans, 14 rue d’Issoudun, P.O. Box 6744, FR-45067 Orléans Cedex 02, France 2 Institute of Applied Material Science, Vietnam Academy of Science and Technology, 1 Mac Dinh Chi, HCMC, Vietnam 3 Sorbonne Universités, UPMC Paris 6, Institut Jean Le Rond d’Alembert, CNRS UMR 7190, 2 place de la gare de ceinture, FR-78210 Saint Cyr l’école, France Abstract: A pulsed non-thermal plasma and 1 wt% Pd/γ-Al2O3 catalyst was used to investigate the CH4, C3H6, and C7H8 oxidation in air. Effects of temperature and specific input energy on the VOCs conversion were studied. The plasma-catalyst interaction revealed the benefit effect on the VOCs oxidation even at low temperature leading to high CO2 selectivity. The synergistic effect of combining plasma with catalyst in one-stage configuration is observed only for toluene. Keywords: non-thermal plasma, Pd/γ-Al203, synergistic effect, methane, propene, toluene, oxidation 1. Introduction 2. Experimental Volatile organic compounds (VOCs) emitted from The plasma reactor is a cylindrical DBD gives the various industrial and domestic processes are important possibility to combine the catalyst with plasma reactor in sources of air pollution and therefore, become a serious single stage (IPC) or two-stage (PPC) configuration as problem for damaging the human health and the shown in Fig. -

HPPO Process Technology a Novel Route to Propylene Oxide Without Coproducts
sustainability/ Industry perspective GREEN CHEMISTRy FRANZ SCHMIDT, MAIK BERNHARD, HEIKO MORELL, MATTHIAS PASCALy* *Corresponding author Evonik Industries AG, Advanced Intermediates – Innovation, Rodenbacher Chaussee 4, 63457 Hanau-Wolfgang, Germany Matthias Pascaly HPPO Process Technology A novel route to propylene oxide without coproducts KEYWORDS: HPPO process, propylene oxide, hydrogen peroxide, titanium silicalite. The common industrial technologies for the conversion of propylene to propylene oxide have been Abstract compared with a special focus on the direct oxidation using hydrogen peroxide. The HPPO process is an economically and ecologically superior technology since there are no market dependencies of other coproducts and water is the only waste product. The catalyst used in this process is a partly titanium substituted silica based zeolite called TS-1. The article summarizes the most important information concerning the HPPO process. INTRODUCTION approximately 7 Mt/a in the year 2010 at a production capacity of approximately 8 Mt/a. Based on a total The oxidation of organic compounds is of vital importance market growth of around 5 % per year, the expected for the chemical industry. Besides basic oxidation reactions values for demand and capacity in 2015 are nearly such as bleaching processes (e.g. paper or laundry) also at 9 and 10 Mt/a respectively. The growing markets are in oxidation reactions of chemicals are important: Epoxides Asia and potentially in the Middle East (1). - especially ethylene and propylene oxide – are among the There are several industrial routes to produce propylene major chemicals. Replacement of traditionally used halide- oxide, of which the chlorohydrin process (CH) is the based oxidants (chlorine) by hydrogen peroxide provided a oldest one (2). -

Process for Preparing Phenol and Acetone from Cumene
Europaisches Patentamt 0 1 64 568 J) European Patent Office Publication number: B1 Office europeen des brevets EUROPEAN PATENT SPECIFICATION Date of publication of patent specification: 06.07.88 (g) int. ci.4: C 07 C 45/53, C 07 C 37/08, C 07 C 27/00, C 07 C 39/04, Application number: 85105584.8 C 07 C 49/08 Date of filing: 07.05.85 Process for preparing phenol and acetone from cumene. (§) Priority: 24.05.84 US 614124 Proprietor: GENERAL ELECTRIC COMPANY 1 River Road Schenectady New York 12305 (US) Date of publication of application: 18.12.85 Bulletin 85/51 Inventor: Fulmer, John William 78 Park Ridge Drive Publication of the grant of the patent: Mt. Vernon Indiana 47620 (US) 06.07.88 Bulletin 88/27 Representative: Catherine, Alain Designated Contracting States: General Electric France Service de Propriete DEFRGBIT Industrielle 18 Rue Horace Vernet B.P. 76 F-92134 Issy-les-Moulineaux Cedex (FR) References cited: EP-A-0 008 869 EP-A-0 028 522 EP-A-0028910 EP-A-0 032 255 AT-B-205490 AT-B- 219 029 00 DE-A-2 756026 DE-A-3 222 533 US-A-2715145 US-A-4480134 <0 Note: Within nine months from the publication of the mention of the grant of the European patent, any person may give notice to the European Patent Office of opposition to the European patent granted. Notice of opposition shall shall be deemed have been filed until the opposition fee has been Q. be filed in a written reasoned statement. -
![Me2si(Benz[E]Indenyl)](https://docslib.b-cdn.net/cover/8000/me2si-benz-e-indenyl-1358000.webp)
Me2si(Benz[E]Indenyl)
Propene Polymerization Using Homogeneous MAO-Activated Metallocene Catalysts: Me2Si(Benz[e]lndenyI)2ZrClu'MAO vs. Me2Si(2-Me-Benz[e]lndenyI)2ZrClu'MAO STEPHAN JUNGLlNG,t ROLF MULHAUPT,b UDO STEHLlNG/ HANS-HERBERT BRINTZINGER/ DAVID FISCHER/ and FRANZ LANGHAUSER3 'Institut fOr Makromolekulare Chemie and Freiburger Materialforschungszentrum der Albert-Ludwigs-Universitat Freiburg, 0-79104 Freiburg, Germany; 2Fakultat fOr Chemie, Universitat Konstanz, 0-78434 Konstanz, Germany; and 3BASF AG Abteilung ZKP, 0-67056 Ludwigshafen, Germany SYNOPSIS Propene was polymerized at 40°C and 2-bar propene in toluene using methylalumoxane (MAO) activated rac-Me2Si(Benz[elIndenyl)2ZrCI2 (BI) and rac-Me2Si(2-Me Benz[elIndenyl)2ZrCl2 (MBI). Catalyst BI/MAO polymerizes propene with high activity to afford low molecular weight polypropylene, whereas MBI/MAO is less active and produces high molecular weight polypropylene. Variation of reaction conditions such as propene concentration, temperature, concentration of catalyst components, and addition of hydrogen reveals that the lower molecular weight polypropylene produced with BI/MAO results from chain transfer to propene monomer following a 2,1-insertion. A large fraction of both me tallocene catalyst systems is deactivated upon 2,1-insertion. Such dormant sites can be reactivated by H2-addition, which affords active metallocene hydrides. This effect of H2- addition is reflected by a decreasing content of head-to-head enchainment and the formation of polypropylene with n-butyl end groups. Both catalysts show a strong dependence of activity on propene concentration that indicates a formal reaction order of 1. 7 with respect to propene. MBI/MAO shows a much higher dependence of the activity on temperature than BI/MAO. -

Propylene Oxide
This report contains the collective views of an in- ternational group of experts and does not necessarily epresent the decisions or the stated policy of the United Nations Environment Programme, the Interna- tional Labour Organisation, or the World Health lOrganization Environmental Health Criteria 56 PROPYLENE OXIDE Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Organization --'-- Geneva, 1985 The International Programme on Chemical Safety (IPCS) is a joint venture the United Nations Environment Programme, the International Labour Organis tion, and the World Health Organization. The main objective of the IPCS is carry Out and disseminate evaluations of the effects of chemicals on human hea' and the quality of the environment. Supporting activities include the developm of epidemiological, experimental laboratory, and risk-assessment methods that cou produce internationally comparable results, and the development of manpower the field of toxicology. Other activities carried out by 1PCS include the develo ment of know-how for coping with chemical accidents, coordination of laborato. testing and epidemiological studies, and promotion of research on the mechanisi of th biological action of chemicals ISBN 92 4 154196 2 \Vorld Health Organization 1985 Publications of the World Health Organization enjoy copyright protection in accordance with the provisions of Protocol 2 of the Universal Copyright Conven- tion. For rights