The Preparation and Hydration Op Unsymmetrical
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Nanoporous Metal-Organic Framework Cu2(BDC)2(DABCO) As an E Cient Heterogeneous Catalyst for One-Pot Facile Synthesis of 1,2,3-T
Scientia Iranica C (2019) 26(3), 1485{1496 Sharif University of Technology Scientia Iranica Transactions C: Chemistry and Chemical Engineering http://scientiairanica.sharif.edu Nanoporous metal-organic framework Cu2(BDC)2(DABCO) as an ecient heterogeneous catalyst for one-pot facile synthesis of 1,2,3-triazole derivatives in ethanol: Evaluating antimicrobial activity of the novel derivatives H. Tourani, M.R. Naimi-Jamal, L. Panahi, and M.G. Dekamin Research Laboratory of Green Organic Synthesis & Polymers, Department of Chemistry, Iran University of Science and Technology, Tehran, P.O. Box 16846-13114, I.R. Iran. Received 6 April 2018; received in revised form 11 August 2018; accepted 31 December 2018 KEYWORDS Abstract. Solvent-free ball-milling synthesized porous metal-organic framework Cu (BDC) (DABCO) (BDC: benzene-1,4-dicarboxylic acid, DABCO: 1,4-diazabicyclo Triazoles; 2 2 [2.2.2]octane) has been proved to be a practical catalyst for facile and convenient synthesis Heterogeneous of 1,2,3-triazole derivatives via multicomponent reaction of terminal alkynes, benzyl or alkyl catalysis; halides, and sodium azide in ethanol. Avoidance of usage and handling of hazardous organic Cu-MOF; azides, using ethanol as an easily available solvent, and simple preparation and recycling Click chemistry; of the catalyst make this procedure a truly scale-up-able one. The high loading of copper Antimicrobial. ions in the catalyst leads to ecient catalytic activity and hence, its low-weight usage in reaction. The catalyst was recycled and reused several times without signi cant loss of its activity. Furthermore, novel derivatives were examined to investigate their potential antimicrobial activity via microdilution method. -

Reactions of Benzene & Its Derivatives
Organic Lecture Series ReactionsReactions ofof BenzeneBenzene && ItsIts DerivativesDerivatives Chapter 22 1 Organic Lecture Series Reactions of Benzene The most characteristic reaction of aromatic compounds is substitution at a ring carbon: Halogenation: FeCl3 H + Cl2 Cl + HCl Chlorobenzene Nitration: H2 SO4 HNO+ HNO3 2 + H2 O Nitrobenzene 2 Organic Lecture Series Reactions of Benzene Sulfonation: H 2 SO4 HSO+ SO3 3 H Benzenesulfonic acid Alkylation: AlX3 H + RX R + HX An alkylbenzene Acylation: O O AlX H + RCX 3 CR + HX An acylbenzene 3 Organic Lecture Series Carbon-Carbon Bond Formations: R RCl AlCl3 Arenes Alkylbenzenes 4 Organic Lecture Series Electrophilic Aromatic Substitution • Electrophilic aromatic substitution: a reaction in which a hydrogen atom of an aromatic ring is replaced by an electrophile H E + + + E + H • In this section: – several common types of electrophiles – how each is generated – the mechanism by which each replaces hydrogen 5 Organic Lecture Series EAS: General Mechanism • A general mechanism slow, rate + determining H Step 1: H + E+ E El e ctro - Resonance-stabilized phile cation intermediate + H fast Step 2: E + H+ E • Key question: What is the electrophile and how is it generated? 6 Organic Lecture Series + + 7 Organic Lecture Series Chlorination Step 1: formation of a chloronium ion Cl Cl + + - - Cl Cl+ Fe Cl Cl Cl Fe Cl Cl Fe Cl4 Cl Cl Chlorine Ferric chloride A molecular complex An ion pair (a Lewis (a Lewis with a positive charge containing a base) acid) on ch lorine ch loronium ion Step 2: attack of -

Acetophenone Sigma Aldrich.Pdf
SIGMA-ALDRICH sigma-aldrich.com Material Safety Data Sheet Version 4.0 Revision Date 02/27/2010 Print Date 07/08/2011 1. PRODUCT AND COMPANY IDENTIFICATION Product name : Acetophenone Product Number : A10701 Brand : Sigma-Aldrich Company : Sigma-Aldrich 3050 Spruce Street SAINT LOUIS MO 63103 USA Telephone : +1 800-325-5832 Fax : +1 800-325-5052 Emergency Phone # : (314) 776-6555 2. HAZARDS IDENTIFICATION Emergency Overview OSHA Hazards Combustible Liquid, Harmful by ingestion., Irritant GHS Label elements, including precautionary statements Pictogram Signal word Danger Hazard statement(s) H227 Combustible liquid H302 Harmful if swallowed. H318 Causes serious eye damage. Precautionary statement(s) P210 Keep away from heat/sparks/open flames/hot surfaces. - No smoking. P264 Wash skin thoroughly after handling. P270 Do not eat, drink or smoke when using this product. P280 Wear protective gloves/protective clothing/eye protection/face protection. P301 + P312 IF SWALLOWED: Call a POISON CENTER or doctor/physician if you feel unwell. P305 + P351 + P338 IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continue rinsing. P310 Immediately call a POISON CENTER or doctor/physician. P330 Rinse mouth. P370 + P378 In case of fire: Use dry sand, dry chemical or alcohol-resistant foam for extinction. P403 + P235 Store in a well-ventilated place. Keep cool. P501 Dispose of contents/container to an approved waste disposal plant. HMIS Classification Health hazard: 2 Flammability: 2 Physical hazards: 0 NFPA Rating Health hazard: 2 Fire: 2 Reactivity Hazard: 0 Potential Health Effects Sigma-Aldrich - A10701 Page 1 of 6 Inhalation May be harmful if inhaled. -

NIPPON SHOKUBAI Company Profile
COMPANY PROFILE Osaka Office Kogin Bldg., 4-1-1 Koraibashi, Chuo-ku, Osaka 541-0043, Japan TEL +81-6-6223-9111 FAX +81-6-6201-3716 Tokyo Office Hibiya Dai Bldg., 1-2-2 Uchisaiwai-cho, Chiyoda-ku, Tokyo 100-0011, Japan TEL +81-3-3506-7475 FAX +81-3-3506-7598 https://www.shokubai.co.jp/en/ 2021.6.2000 Nippon Shokubai Group Mission Management Commitment We are deeply dedicated to humanity and the innate human values of sincerity We conduct all of our corporate and honesty. We respect the unique traits and worldview of each individual, embracing diversity and working to promote mutual understanding and trust. activities based upon a deep We recognize that it is the human spirit and point of view that shape our respect for humanity. understanding of management and our actions. A deep respect for humanity is Providing affluence and comfort to people and society, the foundation for all of our corporate activities. with our unique technology We aim at coexisting with We work to create sustainable societies. We believe it is our corporate social society, and working in responsibility to develop positive relationships with all of our stakeholders, as harmony with the environment. well as with the global environment. We pursue technologies We deliver new value that benefits people and society and are dedicated to working as a unified team to develop technology that will open the door to Corporate Credo that will create the future. the future. Safety takes priority over production. By working to expand our business worldwide, we aim to realize our We act on the global stage. -

ACETOPHENONE Method No. PV2003 Target Concentration
ACETOPHENONE Method no. PV2003 Target concentration: 100 ppm (491 mg/m3) Procedure: Samples are collected by drawing a known volume of air through a Tenax GC tube. Samples are desorbed with a solvent mixture of (5:95) Isopropanol:Carbon Disulfide (IPA):(CS2) and analyzed by gas chromatography with a flame ionization detector. Recommended air volume 120 minutes at 0.1 L/min (12 L) and sampling rate: Status of Method: Partially Validated. This method has been only partially evaluated and is presented for information and trial use. July 1982 Wayne Potter Service Branch 1 OSHA Salt Lake Technical Center Salt Lake City UT-84115 1 of 8 1 General Discussion 1.1 Background 1.1.1 History of procedure Recently, the OSHA Analytical Laboratory received a set of field samples that required analysis for acetophenone. The air samples had been collected on charcoal tubes and isopropanol impingers. Desorption studies were done on charcoal tubes using acetone, carbon disulfide, 5:95 isopropanol:carbon disulfide, and methylene chloride as desorbing solvents. The best results were obtained with 5:95 isopropanol:carbon disulfide, but the recovery was only 68%, which indicated that charcoal tubes should not be recommended for the collection of acetophenone air samples. An evaluation of isopropanol impingers was not performed because it was preferable to find an adsorbent procedure for sample collection. NIOSH method 291 for α -chloroacetophenone uses Tenax GC tubes. The Tenax GC tubes were tried for acetophenone, and appeared to be a suitable method of collection. 1.1.2 Toxic effects (This section is for information only and should not be taken as the basis of OSHA policy.) Acetophenone is a narcotic in high concentrations (Ref. -
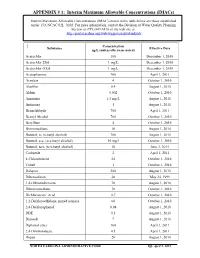
APPENDIX # 1: Interim Maximum Allowable Concentrations (Imacs)
APPENDIX # 1: Interim Maximum Allowable Concentrations (IMACs) Interim Maximum Allowable Concentrations (IMAC) shown in the table below are those established under 15A NCAC 02L .0202. For more information, contact the Division of Water Quality Planning Section at (919) 807-6416 or the web site at: http://portal.ncdenr.org/web/wq/ps/csu/gwstandards Concentration Substance Effective Date ug/L (unless otherwise noted) Acetochlor 100 December 1, 2010 Acetochlor ESA 1 mg/L December 1, 2010 Acetochlor OXA 1 mg/L December 1, 2010 Acetophenone 700 April 1, 2011 Acrolein 4 October 1, 2010 Alachlor 0.4 August 1, 2010 Aldrin 0.002 October 1, 2010 Ammonia 1.5 mg/L August 1, 2010 Antimony 1 August 1, 2010 Benzaldehyde 700 April 1, 2011 Benzyl Alcohol 700 October 1, 2010 Beryllium 4 October 1, 2010 Bromomethane 10 August 1, 2010 Butanol, n- (n-butyl alcohol) 700 August 1, 2010 Butanol, sec- (sec-butyl alcohol) 10 mg/l October 1, 2010 Butanol, tert- (tert-butyl alcohol) 10 June 1, 2011 Carbazole 2 April 1, 2011 4-Chlorotoluene 24 October 1, 2010 Cobalt 1 October 1, 2010 Dalapon 200 August 1, 2010 Dibenzofuran 28 May 24, 1999 1,4-Dibromobenzene 70 August 1, 2010 Dibromomethane 70 October 1, 2010 Dichloroacetic Acid 0.7 October 1, 2010 1,2-Dichloroethylene, mixed isomers 60 October 1, 2010 2,4-Dichlorophenol 0.98 August 1, 2010 DDE 0.1 August 1, 2010 Dinoseb 7 August 1, 2010 Diphenyl ether 100 April 1, 2011 2,4-Dinitrotoluene 0.1 April 1, 2011 Diquat 20 August 1, 2010 NORTH CAROLINA ADMINISTRATIVE CODE Eff. -

Faraday Discussions Accepted Manuscript
View Article Online View Journal Faraday Discussions Accepted Manuscript This manuscript will be presented and discussed at a forthcoming Faraday Discussion meeting. All delegates can contribute to the discussion which will be included in the final volume. Register now to attend! Full details of all upcoming meetings: http://rsc.li/fd-upcoming-meetings This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. Faraday You can find more information about Accepted Manuscripts in the Discussions Information for Authors. Please note that technical editing may introduce minor changes Royal Society of to the text and/or graphics, which may alter content. The journal’s Chemistry standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. This article can be cited before page numbers have been issued, to do this please use: P. Seavill, K. B. Holt and J. D. Wilden, Faraday Discuss., 2019, DOI: 10.1039/C9FD00031C. www.rsc.org/faraday_d Page 1 of 17 Faraday Discussions Investigations Into the Mechanism of Copper-Mediated Glaser-Hay View Article Online Couplings Using Electrochemical Techniques. -

United States Patent Office Patented Nov
3,065,250 United States Patent Office Patented Nov. 20, 1962 i 2 3,065,250 stable in air, becoming brown immediately upon contact NTRLE-METAL CARBONY, COMPLEXES with air. Dewey R. Levering, Wilisagton, Rel, assignor to Her A sample of the product was analyzed under an inert cales Powder Company, Winnington, Del, a corpora atmosphere for percent carbon, hydrogen, nitrogen and tio of Delaware molybdenum. The results of the analysis compared with No Brawing. Filled May 13, 1960, Ser. No. 28,841 the theoretical percentages for (CH3CN)Mo(CO)3 are 26 Caistas. (C. 260-429) tabulated below. The present invention relates to new and useful nitrile metal carbonyl complexes and to the method of their 10 Found ''neory preparation. More specifically, the invention relates to Percent C------------------------------------- 35.4 35.56 nitrile-metal carbonyl complexes where the metal is a Percent H. 2.4 3.0 group VI-B metal (chromium, molybdenum or tungsten) Percent N- 12.35 13.92 according to the periodic system (see Lange's Handbook Percent Mo- 32.6 31.65 of Chemistry, eighth edition, pages 56–57, 1952). Some hydrogen cyanide-metal carbonyl complexes have 5 The product was identified as (CH3CN)Mo(CO)3 been reported. However, complexes of hydrogen cyanide which is in agreement with the evolution of three moles and a group VI-B metal carbonyl have never been re of carbon monoxide per mole of molybdenum carbonyl. ported, and attempts to prepare them have been unsuc The product was insoluble in benzene, carbon tetra cessful. The present invention is based on the unforeseen 20 chloride, and water; somewhat soluble in methanol; and discovery that nitriles undergo a general reaction with soluble in acetonitrile, ethylene glycol dimethyl ether, group VI-B metal carbonyls to form new and useful tetrahydrofuran and dimethylformamide. -

Benzonitrile, 97% MSDS# 98615 Section 1
Material Safety Data Sheet 3,5-Bis(trifluoromethyl)benzonitrile, 97% MSDS# 98615 Section 1 - Chemical Product and Company Identification MSDS Name: 3,5-Bis(trifluoromethyl)benzonitrile, 97% Catalog Numbers: AC311750000, AC311750010 Synonyms: Acros Organics BVBA Company Identification: Janssen Pharmaceuticalaan 3a 2440 Geel, Belgium Acros Organics Company Identification: (USA) One Reagent Lane Fair Lawn, NJ 07410 For information in the US, call: 800-ACROS-01 For information in Europe, call: +32 14 57 52 11 Emergency Number, Europe: +32 14 57 52 99 Emergency Number US: 201-796-7100 CHEMTREC Phone Number, US: 800-424-9300 CHEMTREC Phone Number, Europe: 703-527-3887 Section 2 - Composition, Information on Ingredients ---------------------------------------- CAS#: 27126-93-8 Chemical Name: 3,5-Bis(trifluoromethyl)benzonitrile %: 97 EINECS#: 248-240-7 ---------------------------------------- Hazard Symbols: XN Risk Phrases: 20/21/22 Section 3 - Hazards Identification EMERGENCY OVERVIEW Warning! Combustible liquid and vapor. Harmful if swallowed, inhaled, or absorbed through the skin. Target Organs: No data found. Potential Health Effects Eye: May cause eye irritation. Skin: No information regarding skin irritation and other potential effects was found. Ingestion: The toxicological properties of this substance have not been fully investigated. Inhalation: The toxicological properties of this substance have not been fully investigated. Chronic: Section 4 - First Aid Measures Flush eyes with plenty of water for at least 15 minutes, occasionally lifting the upper and lower eyelids. Get Eyes: medical aid immediately. Get medical aid immediately. Flush skin with plenty of water for at least 15 minutes while removing Skin: contaminated clothing and shoes. If victim is conscious and alert, give 2-4 cupfuls of milk or water. -

DESIGN, SYNTHESIS, CHARACTERIZATION of SOME NEW SUBSTITUTED CHALCONES and STUDIES THEIR ANTIMICROBIAL ACTIVITIES Zakaria H
[Aiube* et al., 5(6): June, 2016] ISSN: 2277-9655 IC™ Value: 3.00 Impact Factor: 4.116 IJESRT INTERNATIONAL JOURNAL OF ENGINEERING SCIENCES & RESEARCH TECHNOLOGY DESIGN, SYNTHESIS, CHARACTERIZATION OF SOME NEW SUBSTITUTED CHALCONES AND STUDIES THEIR ANTIMICROBIAL ACTIVITIES Zakaria H. Aiube*, Ali H. Samir, Israa SH. A- R. Al- Kadi * Department of Chemistry, college of Education for pure Science-Ibn, Al- Haitham, University, Baghdad, Iraq DOI: 10.5281/zenodo.55982 ABSTRACT Eight designed chalcones, named [1(p-benzenesulphonamidophenyl)-3-p-chloro-2-propene-1-one][2], [1(p- benzenesulphonamidophenyl)-3-p-nitro-2-propene-1-one][3], [1(4-Ureido)phenyl-3-p-chlorophenyl-2-propene- 1-one][5], [1(4-Ureido)phenyl-3-p-nitroophenyl-2-propene-1-one][6], [1(4(p-N-methylaminophenyl)azophenyl- 3-p-chlorophenyl-2-propene-1-one][8], [1(4(p-N-methylaminophenyl)azophenyl-3-p-nitroophenyl-2-propene-1- one][9], [1(p-aminophenyl)-3-p-chlorophenyl-2-propene-1-one][10] and [1(p-aminophenyl)-3-p-nitrophenyl-2- propene-1-one][11], were synthesised by condensation of synthesised p-acetylphenylbenzene- sulphonamide, p- acetylphenylurea and p-acetyl-p'-(N-methylamino)azobenzene, with p-chlorobenzaldehyde and p nitrobenzaldehyde in basic media respectively. All synthesised compounds are characterized by its melting points, FTIR, 1H NMR, 13C NMR and Mass spectral analysis. All synthesised compounds are examined their antimicrobial activities against Gram-Ve bactria (Serratia marcescens, Pseudmonas aeroginosa) and Gram+Ve bacterial (Stphylococcus aureus, Streptococcus pyogenes), and Candida albicans fungi. Result showed good to moderate inhibition effect against some bacteria and fungi, in comparison with some pharmaceutical antibiotic and antifungal treatments like Cephalexin, Amoxicillin, Tetracycline, Lincomycine, Nystatine and Fluconazole respectively. -

SYNTHESIS of PERDEUTEROPHENYLACETYLENE Kv Joy T
B.A.fl.C-1332 CD > JO b w U) SYNTHESIS OF PERDEUTEROPHENYLACETYLENE kv Joy T. Kunjappu, C. V. C. Prasad, H. M. Gupta, M. H. Rao and K. N. Rao Chemistry Division 1986 B.A.R.C. - 1332 GOVERNMENT OP INDIA ATOMIC ENERGY COMMISSION * SYNTHESIS OF PERDEUTEROPHENYLACETYLENE by Joy T. Kunjappu, C.V.C. ^rasad, H.M. Gupta, M.H. Rao and K.N. Rao Chemistry Division BHABHA ATOMIC RESEARCH CENTRE BOMBAY, INDIA 1986 BARC . 1332 INIS Subject Category J B12. 10 Descriptor a ACETYLENES DEUTERATION DEUTERIUM COMPOUNDS CHEMICAL PREPARATION TARGETS MATERIALS THERMONUCLEAR REACTIONS INFRARED SPECTRA QUANTITATIVE CHEMICAL ANALYSIS Abstract The relative merits of feasible methods to synthesise phenyl acetylene -d have been evaluated and a convenient path has been adopted for obtaining it in good isotoplc purity. Detailed procedures for preparing the intermediate deuterated compounds like benzene- bromobenzene-d , o£-phenyl-d -ethanol, phenyl-d -ethylene, styrene 5 5 6 dibromide -d , phenyl-d -acetylene, phenyl acetylene-d and deutero- sulphuric acid have been outlined, The infrared spectra of these deutero compounds in carbon tetrachlorlde solution scanned In sodium chloride liquid cells have been presented and they were used in quantitatively estimating their deuterium content. SYNTHESIS OF PERDEUTEROPHENYLACETYLENE: Joy T. Kunjappu, C.V.C. Prasad, H.M. Gupta, M.H. Rao and K.N. Rao A. Introduction: Deuterated organic compounds find application in nuclear and allied fields . They are also useful in mechanistic studies' ' employed in ascertaining the Identity of chemical bonds involved In chemical reactions, by following isotopic kinetic effects and other related methods. Both monomerlc and polymeric deutero compounds have been useful in nuclear related studies. -

Selective Reduction of Aromatic Nitriles to Aldehydes by Lithium Diisobutylpiperidinohydroaluminate (LDBPA)
Notes Bull. Korean Chem. Soc. 2006, Vol. 27, No. 1 121 Selective Reduction of Aromatic Nitriles to Aldehydes by Lithium Diisobutylpiperidinohydroaluminate (LDBPA) Ji Hyun Ha, Jin Hee Ahn; and Duk Keun An* Department of Chemistry, Kangwon National University, Chuncheon 200-701, Korea. *E-mail: dkan@kangwon. ac. kr ^Medicinal Science Division, Korea Research Institute of Chemical Technology, Yusung, Daejeon 305-600, Korea Received October 24, 2005 Key Words : Lithium diisobutylpiperidinohydroaluminate (LDBPA), Diisobutylaluminium hydride (DIBAH), Aldehyde, Nitriles, Piperidine Partial reduction of nitriles into aldehyde is one of the As a part of our research program directed toward the most important and highly desirable means in organic discovery of new reducing agents through the simple synthesis, and a large number of reducing agents for this modification of commercial DIBAH (Scheme 1), we applied have been reported.1 Among them, a commercially available this reagent for the partial reduction of nitriles to aldehydes. diisobutylaluminium hydride (DIBAH)11 is commonly used We first examined partial reduction of benzonitrile and for the preparation of aldehydes from both aromatic and capronitrile with 1 in THF at 0 °C. The reduction of aliphatic nitriles, although the reagent provides moderate benzonitrile provided benzaldehyde in quantitative yield in 1 yields (48-90%). It has been also reported that other several h. Using the same methodology, the partial reduction of reducing agents such as potassium 9-5,ec-amyl-9-borata- other aromatic and aliphatic nitriles to the corresponding cycl이 3.3.1]nonane (K-9-.vec-Am-9-BBNH),lf lithium tris- aldehydes was carried out. As shown in Table 1, aromatic (dihexylamino)alummium hydride (LTDHA)lg and lithium benzonitriles, such as 1 -cyanonaphthalene, chlorobenzo JV;JV-dimethylethylenediammoalummium hydride (LDME- nitriles, toluonitriles and methoxybenzonitriles were smooth DAH)lk reduce chemoselectively nitriles to aldehydes.