Discovery, Expression Profiling, and Evolutionary Analysis Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ISTA List of Stabilized Plant Names 7Th Edition
ISTA List of Stabilized Plant Names th 7 Edition ISTA Nomenclature Committee Chair: Dr. M. Schori Published by All rights reserved. No part of this publication may be The Internation Seed Testing Association (ISTA) reproduced, stored in any retrieval system or transmitted Zürichstr. 50, CH-8303 Bassersdorf, Switzerland in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior ©2020 International Seed Testing Association (ISTA) permission in writing from ISTA. ISBN 978-3-906549-77-4 ISTA List of Stabilized Plant Names 1st Edition 1966 ISTA Nomenclature Committee Chair: Prof P. A. Linehan 2nd Edition 1983 ISTA Nomenclature Committee Chair: Dr. H. Pirson 3rd Edition 1988 ISTA Nomenclature Committee Chair: Dr. W. A. Brandenburg 4th Edition 2001 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 5th Edition 2007 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 6th Edition 2013 ISTA Nomenclature Committee Chair: Dr. J. H. Wiersema 7th Edition 2019 ISTA Nomenclature Committee Chair: Dr. M. Schori 2 7th Edition ISTA List of Stabilized Plant Names Content Preface .......................................................................................................................................................... 4 Acknowledgements ....................................................................................................................................... 6 Symbols and Abbreviations .......................................................................................................................... -

Invasive Plants in Southern Forests
Invasive Plants in Southern Forests United States Department of Agriculture A Field Guide for the Identification of Invasive PlantsSLIGHTLY inREVISED NOVEMBERSouthern 2015 Forests United States Forest Service Department Southern Research Station James H. Miller, Erwin B. Chambliss, and Nancy J. Loewenstein of Agriculture General Technical Report SRS–119 Authors: James H. Miller, Emeritus Research Ecologist, and Erwin B. Chambliss, Research Technician, Forest Available without charge from the Service, U.S. Department of Agriculture, Southern Research Station, Auburn University, AL 36849; and Southern Research Station Nancy J. Loewenstein, Research Fellow and Alabama Cooperative Extension System Specialist for Also available online at Forest Invasive Plants, School of Forestry and Wildlife Sciences, Auburn University, AL 36849. www.srs.fs.usda.gov/pubs/35292 and invasive.org, or as a free download for iPhones and iPads at the AppStore Front Cover Upper left—Chinese lespedeza (Lespedeza cuneata) infestation that developed from dormant seed in the soil seed bank after a forest thinning operation. Upper right—Kudzu (Pueraria montana) infestation within the urban-wildland interface. Lower left—Chinese privet (Ligustrum sinense) and dormant kudzu invading and replacing a pine- hardwood stand. Lower right—Cogongrass (Imperata cylindrica) infestation under mature slash pine (Pinus elliottii). Funding support for all printings provided by the Southern Research Station, Insect, Disease, and Invasive Plants Research Work Unit, and Forest Health Protection, Southern Region, Asheville, NC. First Printed April 2010 Slightly Revised February 2012 Revised August 2013 Reprinted January 2015 Slightly Revised November 2015 Southern Research Station 200 W.T. Weaver Blvd. Asheville, NC 28804 www.srs.fs.usda.gov i A Field Guide for the Identification of Invasive Plants in Southern Forests James H. -

Elymus Elymoides Crested Wheatgrass Agropyron Desertorum
Reclamation of Pipeline Right-Of-Ways on Rangelands Charlie D. Clements, James A. Young and Dan N. Harmon Introduction USDA, Agricultural Research Service 920 Valley Rd. Reno NV 89512 [email protected] In the mid 1990’s a large diameter natural gas pipeline was constructed across western Nevada. Reclamation of such right-of-ways are challenging because of both the severity and the linear nature of the disturbance. During construction, pipelines and equipment (Fig. 1) are highly visible at a landscape level to the general public, therefore attracting concerns and reclamation suggestions from a host of interest groups. The construction contractor invited us to set up plots and conduct research concerning the reclamation of their pipeline disturbance. Table 1. Plant material used in the pipeline reclamation effort. Common Name _____ Scientific Name Antelope Bitterbrush Purshia tridentata Figure 3. Lower pipeline site 6 years after the initial reclamation seeding. Basin Big Sagebrush Artemisia tridentata Basin Wildrye Leymus cinereus Big Bluegrass Poa secunda Bluebunch Wheatgrass Pseudoroegneria spicata Figure 1. Heavy equipment used in pipeline construction is very visible as well as the disturbance. Bottlebrush Squirreltail Elymus elymoides Crested Wheatgrass Agropyron desertorum Desert Needlegrass Achnatherum speciosum Idaho Fescue Festuca idahoensis ‘Immigrant’ Forage Kochia Kochia prostrata Indian Ricegrass Achnatherum hymenoides Needle-and-Threadgrass Hesperostipa comata Shadscale Atriplex confortifolia Thurber’s Needlegrass Achnatherum thurberianum Figure 4. Antelope bitterbrush, basin big sagebrush, ‘Immigrant’ forage kochia,and needle-and-thread grass were some of the planted species that successfully established. Figure 2. Pipeline site the first year after reclamation seeding effort. Results and Discussion It is a common practice to rest sites from livestock grazing for 2 years following such efforts, but Methods this is hard to do following pipeline disturbances because the disturbances are so narrow and linear in nature. -

(12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer Et Al
USOO8440393B2 (12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer et al. (45) Date of Patent: May 14, 2013 (54) PRO-ANGIOGENIC GENES IN OVARIAN OTHER PUBLICATIONS TUMORENDOTHELIAL CELL, SOLATES Boyd (The Basic Science of Oncology, 1992, McGraw-Hill, Inc., p. (75) Inventors: Michael J. Birrer, Mt. Airy, MD (US); 379). Tomas A. Bonome, Washington, DC Tockman et al. (Cancer Res., 1992, 52:2711s-2718s).* (US); Anil Sood, Pearland, TX (US); Pritzker (Clinical Chemistry, 2002, 48: 1147-1150).* Chunhua Lu, Missouri City, TX (US) Benedict et al. (J. Exp. Medicine, 2001, 193(1) 89-99).* Jiang et al. (J. Biol. Chem., 2003, 278(7) 4763-4769).* (73) Assignees: The United States of America as Matsushita et al. (FEBS Letters, 1999, vol. 443, pp. 348-352).* Represented by the Secretary of the Singh et al. (Glycobiology, 2001, vol. 11, pp. 587-592).* Department of Health and Human Abbosh et al. (Cancer Res. Jun. 1, 2006 66:5582-55.91 and Supple Services, Washington, DC (US); The mental Figs. S1-S7).* University of MD Anderson Cancer Zhai et al. (Chinese General Practice Aug. 2008, 11(8A): 1366 Center, Houston, TX (US) 1367).* Lu et al. (Cancer Res. Feb. 15, 2007, 64(4): 1757-1768).* (*) Notice: Subject to any disclaimer, the term of this Bagnato et al., “Activation of Mitogenic Signaling by Endothelin 1 in patent is extended or adjusted under 35 Ovarian Carcinoma Cells', Cancer Research, vol. 57, pp. 1306-1311, U.S.C. 154(b) by 194 days. 1997. Bouras et al., “Stanniocalcin 2 is an Estrogen-responsive Gene (21) Appl. -

USSR (North Caucasus, Kazakhstan Republic) July 18-August 31, 1977 U.S
PLANT GERMPLASM COLLECTION REPORT USDA-ARS FORAGE AND RANGE RESEARCH LABORATORY LOGAN, UTAH Foreign Travel to: USSR (North Caucasus, Kazakhstan Republic) July 18-August 31, 1977 U.S. Participants Douglas R. Dewey - Research Geneticist (deceased) contact Jack Staub USDA-Agricultural Research Service Logan, Utah U.S.A. A. Perry Plummer USDA-Forest Service Provo, Utah U.S.A. Laurie Law (Interpreter) USDA-ARS-IPD Washington, DC 20250 GERMPLASM ACCESSIONS Country Visited: Soviet Union -North Caucasus (Stavropol Kray) -Kazakstan Republic (Tselinograd Oblast) (Alma Ata Oblast) (Dzhambul Oblast) (Chimkent Oblast) Period of Travel: July 18 - August 31, 1977 Moscow: July 18-19 Stavropol: July 20 - August 6 Tselinograd: August 7-11 Alma Ata: August 12-16 Dzhambul: August 17-21 Chimkent: August 22-28 Moscow: August 29-31 Purpose of Trip: 1) To collect germplasm of grasses, legumes, forbs, and shrubs from natural large sites in the USSR for possible use on U.S. rangelands; 2) to establish contracts with Soviet botanists and plant breeders for the purpose of negotiating future seed exchanges. SUMMARY A 45-day plant collection expedition to the USSR by D. R. Dewey, A. P. Plummer, and Laurie Law netted about 1,100 seed collections of range-forage grasses, legumes, forbs, and shrubs. The Soviets provided land transportation and an escort of several scientists and administrators throughout the trip. The collectors were usually housed in hotels and made daily trips to collect native vegetation in surrounding areas usually within a 100-km radius. Almost 3 weeks (July 20 - August 6) were spent in the Stavropol Kray in the northern foothills of the Caucasus Mountains. -

Pathogenetic Subgroups of Multiple Myeloma Patients
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector ARTICLE High-resolution genomic profiles define distinct clinico- pathogenetic subgroups of multiple myeloma patients Daniel R. Carrasco,1,2,8 Giovanni Tonon,1,8 Yongsheng Huang,3,8 Yunyu Zhang,1 Raktim Sinha,1 Bin Feng,1 James P. Stewart,3 Fenghuang Zhan,3 Deepak Khatry,1 Marina Protopopova,5 Alexei Protopopov,5 Kumar Sukhdeo,1 Ichiro Hanamura,3 Owen Stephens,3 Bart Barlogie,3 Kenneth C. Anderson,1,4 Lynda Chin,1,7 John D. Shaughnessy, Jr.,3,9 Cameron Brennan,6,9 and Ronald A. DePinho1,5,9,* 1 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts 02115 2 Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts 02115 3 The Donna and Donald Lambert Laboratory of Myeloma Genetics, Myeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205 4 The Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts 02115 5 Center for Applied Cancer Science, Belfer Institute for Innovative Cancer Science, Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts 6 Neurosurgery Service, Memorial Sloan-Kettering Cancer Center, Department of Neurosurgery, Weill Cornell Medical College, New York, New York 10021 7 Department of Dermatology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts, 02115 8 These authors contributed equally to this work. 9 Cocorresponding authors: [email protected] (C.B.); [email protected] (J.D.S.); [email protected] (R.A.D.) *Correspondence: [email protected] Summary To identify genetic events underlying the genesis and progression of multiple myeloma (MM), we conducted a high-resolu- tion analysis of recurrent copy number alterations (CNAs) and expression profiles in a collection of MM cell lines and out- come-annotated clinical specimens. -

Literature Cited Robert W. Kiger, Editor This Is a Consolidated List Of
RWKiger 26 Jul 18 Literature Cited Robert W. Kiger, Editor This is a consolidated list of all works cited in volumes 24 and 25. In citations of articles, the titles of serials are rendered in the forms recommended in G. D. R. Bridson and E. R. Smith (1991). When those forms are abbreviated, as most are, cross references to the corresponding full serial titles are interpolated here alphabetically by abbreviated form. Two or more works published in the same year by the same author or group of coauthors will be distinguished uniquely and consistently throughout all volumes of Flora of North America by lower-case letters (b, c, d, ...) suffixed to the date for the second and subsequent works in the set. The suffixes are assigned in order of editorial encounter and do not reflect chronological sequence of publication. The first work by any particular author or group from any given year carries the implicit date suffix "a"; thus, the sequence of explicit suffixes begins with "b". Works missing from any suffixed sequence here are ones cited elsewhere in the Flora that are not pertinent in these volumes. Aares, E., M. Nurminiemi, and C. Brochmann. 2000. Incongruent phylogeographies in spite of similar morphology, ecology, and distribution: Phippsia algida and P. concinna (Poaceae) in the North Atlantic region. Pl. Syst. Evol. 220: 241–261. Abh. Senckenberg. Naturf. Ges. = Abhandlungen herausgegeben von der Senckenbergischen naturforschenden Gesellschaft. Acta Biol. Cracov., Ser. Bot. = Acta Biologica Cracoviensia. Series Botanica. Acta Horti Bot. Prag. = Acta Horti Botanici Pragensis. Acta Phytotax. Geobot. = Acta Phytotaxonomica et Geobotanica. [Shokubutsu Bunrui Chiri.] Acta Phytotax. -
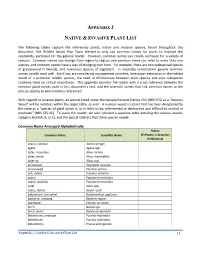
Reference Plant List
APPENDIX J NATIVE & INVASIVE PLANT LIST The following tables capture the referenced plants, native and invasive species, found throughout this document. The Wildlife Action Plan Team elected to only use common names for plants to improve the readability, particular for the general reader. However, common names can create confusion for a variety of reasons. Common names can change from region-to-region; one common name can refer to more than one species; and common names have a way of changing over time. For example, there are two widespread species of greasewood in Nevada, and numerous species of sagebrush. In everyday conversation generic common names usually work well. But if you are considering management activities, landscape restoration or the habitat needs of a particular wildlife species, the need to differentiate between plant species and even subspecies suddenly takes on critical importance. This appendix provides the reader with a cross reference between the common plant names used in this document’s text, and the scientific names that link common names to the precise species to which writers referenced. With regards to invasive plants, all species listed under the Nevada Revised Statute 555 (NRS 555) as a “Noxious Weed” will be notated, within the larger table, as such. A noxious weed is a plant that has been designated by the state as a “species of plant which is, or is likely to be, detrimental or destructive and difficult to control or eradicate” (NRS 555.05). To assist the reader, we also included a separate table detailing the noxious weeds, category level (A, B, or C), and the typical habitats that these species invade. -

ALKALI SACATON (Sporobolus Airoides) Section 7.1.4, US ARMY CORPS of ENGINEERS WILDLIFE RESOURCES MANAGEMENT MANUAL
FlSH & WlLOUFE REFERENCE LIBRARY ENVIRONMENTAL IMPACT RESEARCH PROGRAM • TECHNICAL REPORT EL-86-37 ALKALI SACATON (Sporobolus airoides) Section 7.1.4, US ARMY CORPS OF ENGINEERS WILDLIFE RESOURCES MANAGEMENT MANUAL by Clinton H. Wasser Colorado State University Fort Collins, Colorado 80523 and Phillip L. Dittberner, Donald R. Dietz US Fish and Wildlife Service Fort Collins, Colorado 80526 • July 1986 Final Report Approved For Public Release; Distribution Unlimited Prepared for DEPARTMENT OF THE ARMY US Army Corps of Engineers Washington, DC 20314-1000 Under EIRP Work Unit 31631 Monitored by Environmental Laboratory • US Army Engineer Waterways Experiment Station PO Box 631, Vicksburg, Mississippi 39180-0631 Unclassified SECURITY CLASSIFICATION OF THIS PAGE IForm Approved REPORT DOCUMENTATION PAGE OM8 No. 0704-0188 • Exp. Date: Jun 30, 1986 1a. REPORT SECURITY CLASSIFICATION 1 b. RESTRICTIVE MARKINGS Uncla<:<:ified 2a. SECURITY CLASSIFICATION AUTHORITY 3. DISTRIBUTION I AVAILABILITY OF REPORT Approved for public release; 2b. DECLASSIFICATION I DOWNGRADING SCHEDULE distribution unlimited. 4. PERFORMING ORGANIZATION REPORT NUMBER(S) 5. MONITORING ORGANIZATION REPORT NUMBER(S) Technical Report EL-86-37 6a. NAME OF PERFORMING ORGANIZATION 6b. OFFICE SYMBOL la. NAME OF MONITORING ORGANIZATION See reverse (If applicable) USAEWES Environmental Laboratorv 6c. ADDRESS (City, State, and ZIP Code) 7b. ADDRESS (City, State, and ZIP Code) See reverse PO Box 631 Vicksburg, MS 39180-0631 Ba. NAME OF FUNDING /SPONSORING Sb. OFFICE SYMBOL 9. PROCUREMENT INSTRUMENT IDENTIFICATION NUMBER ORGANIZATION (If applicable) US Army Corps of Engineers Sc. ADDRESS (City, State, and ZIP Code) 10. SOURCE OF FUNDING NUMBERS PROGRAM PROJECT TASK WORK UNIT ELEMENT NO. NO. NO. ACCESSION NO Washington, DC 20314-1000 EIRP 316'H 11. -

Spotted Knapweed
Spotted knapweed (Centaurea maculosa Lam) : water, nutrients, plant competition, bacteria, and the seed head fly (Urophora affinis Frnfd.) by Stephen Anthony Kearing A thesis submitted in partial fulfillment of the requirements for the degree of Master of Science in Entomology Montana State University © Copyright by Stephen Anthony Kearing (1996) Abstract: Spotted knapweed, Centaurea maculosa Lam., is considered fay many to be the number one noxious weed in western Montana. A hydroponics study was conducted in Bozeman, MT to quantify water and nutrient uptake of spotted knapweed and two grass competitors. Spotted knapweed, western wheatgrass, Pascopymm smithii (Rydb.) Love, and a crested wheatgrass hybrid, Agropyron cristatum (L.) Gaert X Agropyron desertorum (Fisch. ex Link) Schult., were grown in a complete randomized block design, each in individual hydroponic drip systems with an inert rock wool media. The pots were weighed at each interval to estimate water use. Mean concentrations remaining in the system for nitrogen, phosphorous, and potassium were regressed against mean cumulative water use for each treatment. Spotted knapweed and western wheatgrass had similar slopes for nitrogen and potassium concentrations that were significantly lower than crested wheatgrass (t-test, P < = 0.05), suggesting that knapweed and western wheatgrass absorb nitrogen and potassium more efficiently than crested wheatgrass. Conversely, crested wheatgrass had a significantly lower slope for concentrations of phosphorous remaining in the solution (t-test, P = 0.01), suggesting crested wheatgrass absorbs more phosphorous per ml water than western wheatgrass or spotted knapweed. Spotted knapweed used more water throughout the experiment (P = 0.01), with differences being greatest during bolting. The combination of water and nutrient uptake rates help to explain spotted knapweed’s ability to compete for resources. -

Susceptibility to Glaucoma: Differential Comparison of the Astrocyte
Open Access Research2008LukasetVolume al. 9, Issue 7, Article R111 Susceptibility to glaucoma: differential comparison of the astrocyte transcriptome from glaucomatous African American and Caucasian American donors Thomas J Lukas¤*, Haixi Miao¤†, Lin Chen†, Sean M Riordan†, Wenjun Li†, Andrea M Crabb†, Alexandria Wise‡, Pan Du§, Simon M Lin§ and M Rosario Hernandez† Addresses: *Department of Molecular Pharmacology and Biological Chemistry, Feinberg School of Medicine, Northwestern University, E Chicago Ave, Chicago, IL 60611 USA. †Department of Ophthalmology, Feinberg School of Medicine, Northwestern University, E Chicago Ave, Chicago, IL 60611 USA. ‡Department of Biology, City College of New York, Convent Ave, New York, NY 10031, USA. §Robert H Lurie Comprehensive Cancer Center, Feinberg School of Medicine, Northwestern University, E Chicago Ave, Chicago, IL 60611 USA. ¤ These authors contributed equally to this work. Correspondence: Thomas J Lukas. Email: [email protected] Published: 9 July 2008 Received: 9 May 2008 Revised: 18 June 2008 Genome Biology 2008, 9:R111 (doi:10.1186/gb-2008-9-7-r111) Accepted: 9 July 2008 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2008/9/7/R111 © 2008 Lukas et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Epidemiological and genetic studies indicate that ethnic/genetic background plays an important role in susceptibility to primary open angle glaucoma (POAG). -

Identification of Dysregulated Genes in Rheumatoid Arthritis Based on Bioinformatics Analysis
Identification of dysregulated genes in rheumatoid arthritis based on bioinformatics analysis Ruihu Hao1,*, Haiwei Du2,*, Lin Guo1, Fengde Tian1, Ning An1, Tiejun Yang3, Changcheng Wang1, Bo Wang1 and Zihao Zhou1 1 Department of Orthopedics, Affiliated Zhongshan Hospital of Dalian University, Dalian, China 2 Department of Bioinformatics, Beijing Medintell Biomed Co., Ltd, Beijing, China 3 Department of Orthopedics, Affiliated Hospital of BeiHua University, Jilin, China * These authors contributed equally to this work. ABSTRACT Background. Rheumatoid arthritis (RA) is a chronic auto-inflammatory disorder of joints. The present study aimed to identify the key genes in RA for better understanding the underlying mechanisms of RA. Methods. The integrated analysis of expression profiling was conducted to identify differentially expressed genes (DEGs) in RA. Moreover, functional annotation, protein– protein interaction (PPI) network and transcription factor (TF) regulatory network construction were applied for exploring the potential biological roles of DEGs in RA. In addition, the expression level of identified candidate DEGs was preliminarily detected in peripheral blood cells of RA patients in the GSE17755 dataset. Quantitative real-time polymerase chain reaction (qRT-PCR) was conducted to validate the expression levels of identified DEGs in RA. Results. A total of 378 DEGs, including 202 up- and 176 down-regulated genes, were identified in synovial tissues of RA patients compared with healthy controls. DEGs were significantly enriched in