Identification of Dysregulated Genes in Rheumatoid Arthritis Based on Bioinformatics Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Eif4gi Cooperation in Start Codon Selection
Article Translational Tolerance of Mitochondrial Genes to Metabolic Energy Stress Involves TISU and eIF1- eIF4GI Cooperation in Start Codon Selection Graphical Abstract Authors Hadar Sinvani, Ora Haimov, ..., Benoit Viollet, Rivka Dikstein Correspondence [email protected] In Brief The regulatory element TISU is a translation initiator of short 50 UTR mRNAs. Sinvani et al. report that TISU is enriched in mitochondrial genes and confers resistance to translation inhibition mediated by AMPK and energy deprivation, allowing cells to cope with the stress. TISU-mediated initiation involves cap complex eviction and eIF1- eIF4G1 cooperation. Highlights 0 d TISU, translation initiator of short 5 UTR, is enriched among mitochondrial genes d TISU-mediated initiation involves eIF1-eIF4G1 cooperation and cap complex eviction d Low energy induces a stress response that includes inhibition of global translation d TISU-directed translation enables continuous translation under energy stress Sinvani et al., 2015, Cell Metabolism 21, 479–492 March 3, 2015 ª2015 Elsevier Inc. http://dx.doi.org/10.1016/j.cmet.2015.02.010 Cell Metabolism Article Translational Tolerance of Mitochondrial Genes to Metabolic Energy Stress Involves TISU and eIF1-eIF4GI Cooperation in Start Codon Selection Hadar Sinvani,1,4 Ora Haimov,1,4 Yuri Svitkin,2 Nahum Sonenberg,2 Ana Tamarkin-Ben-Harush,1 Benoit Viollet,3 and Rivka Dikstein1,* 1Department of Biological Chemistry, The Weizmann Institute of Science, Rehovot 76100, Israel 2Department of Biochemistry and Goodman Cancer Centre, McGill University, Montreal, QC H3A 1A3, Canada 3University Paris Descartes, Institut Cochin, 75014 Paris, France 4Co-first author *Correspondence: [email protected] http://dx.doi.org/10.1016/j.cmet.2015.02.010 SUMMARY 2010; Ja¨ ger et al., 2007; Lin et al., 2005; O’Neill et al., 2011; Winder et al., 2000). -

(12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer Et Al
USOO8440393B2 (12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer et al. (45) Date of Patent: May 14, 2013 (54) PRO-ANGIOGENIC GENES IN OVARIAN OTHER PUBLICATIONS TUMORENDOTHELIAL CELL, SOLATES Boyd (The Basic Science of Oncology, 1992, McGraw-Hill, Inc., p. (75) Inventors: Michael J. Birrer, Mt. Airy, MD (US); 379). Tomas A. Bonome, Washington, DC Tockman et al. (Cancer Res., 1992, 52:2711s-2718s).* (US); Anil Sood, Pearland, TX (US); Pritzker (Clinical Chemistry, 2002, 48: 1147-1150).* Chunhua Lu, Missouri City, TX (US) Benedict et al. (J. Exp. Medicine, 2001, 193(1) 89-99).* Jiang et al. (J. Biol. Chem., 2003, 278(7) 4763-4769).* (73) Assignees: The United States of America as Matsushita et al. (FEBS Letters, 1999, vol. 443, pp. 348-352).* Represented by the Secretary of the Singh et al. (Glycobiology, 2001, vol. 11, pp. 587-592).* Department of Health and Human Abbosh et al. (Cancer Res. Jun. 1, 2006 66:5582-55.91 and Supple Services, Washington, DC (US); The mental Figs. S1-S7).* University of MD Anderson Cancer Zhai et al. (Chinese General Practice Aug. 2008, 11(8A): 1366 Center, Houston, TX (US) 1367).* Lu et al. (Cancer Res. Feb. 15, 2007, 64(4): 1757-1768).* (*) Notice: Subject to any disclaimer, the term of this Bagnato et al., “Activation of Mitogenic Signaling by Endothelin 1 in patent is extended or adjusted under 35 Ovarian Carcinoma Cells', Cancer Research, vol. 57, pp. 1306-1311, U.S.C. 154(b) by 194 days. 1997. Bouras et al., “Stanniocalcin 2 is an Estrogen-responsive Gene (21) Appl. -

Pathogenetic Subgroups of Multiple Myeloma Patients
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector ARTICLE High-resolution genomic profiles define distinct clinico- pathogenetic subgroups of multiple myeloma patients Daniel R. Carrasco,1,2,8 Giovanni Tonon,1,8 Yongsheng Huang,3,8 Yunyu Zhang,1 Raktim Sinha,1 Bin Feng,1 James P. Stewart,3 Fenghuang Zhan,3 Deepak Khatry,1 Marina Protopopova,5 Alexei Protopopov,5 Kumar Sukhdeo,1 Ichiro Hanamura,3 Owen Stephens,3 Bart Barlogie,3 Kenneth C. Anderson,1,4 Lynda Chin,1,7 John D. Shaughnessy, Jr.,3,9 Cameron Brennan,6,9 and Ronald A. DePinho1,5,9,* 1 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts 02115 2 Department of Pathology, Brigham and Women’s Hospital, Boston, Massachusetts 02115 3 The Donna and Donald Lambert Laboratory of Myeloma Genetics, Myeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences, Little Rock, Arkansas 72205 4 The Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts 02115 5 Center for Applied Cancer Science, Belfer Institute for Innovative Cancer Science, Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts 6 Neurosurgery Service, Memorial Sloan-Kettering Cancer Center, Department of Neurosurgery, Weill Cornell Medical College, New York, New York 10021 7 Department of Dermatology, Brigham and Women’s Hospital and Harvard Medical School, Boston, Massachusetts, 02115 8 These authors contributed equally to this work. 9 Cocorresponding authors: [email protected] (C.B.); [email protected] (J.D.S.); [email protected] (R.A.D.) *Correspondence: [email protected] Summary To identify genetic events underlying the genesis and progression of multiple myeloma (MM), we conducted a high-resolu- tion analysis of recurrent copy number alterations (CNAs) and expression profiles in a collection of MM cell lines and out- come-annotated clinical specimens. -

Susceptibility to Glaucoma: Differential Comparison of the Astrocyte
Open Access Research2008LukasetVolume al. 9, Issue 7, Article R111 Susceptibility to glaucoma: differential comparison of the astrocyte transcriptome from glaucomatous African American and Caucasian American donors Thomas J Lukas¤*, Haixi Miao¤†, Lin Chen†, Sean M Riordan†, Wenjun Li†, Andrea M Crabb†, Alexandria Wise‡, Pan Du§, Simon M Lin§ and M Rosario Hernandez† Addresses: *Department of Molecular Pharmacology and Biological Chemistry, Feinberg School of Medicine, Northwestern University, E Chicago Ave, Chicago, IL 60611 USA. †Department of Ophthalmology, Feinberg School of Medicine, Northwestern University, E Chicago Ave, Chicago, IL 60611 USA. ‡Department of Biology, City College of New York, Convent Ave, New York, NY 10031, USA. §Robert H Lurie Comprehensive Cancer Center, Feinberg School of Medicine, Northwestern University, E Chicago Ave, Chicago, IL 60611 USA. ¤ These authors contributed equally to this work. Correspondence: Thomas J Lukas. Email: [email protected] Published: 9 July 2008 Received: 9 May 2008 Revised: 18 June 2008 Genome Biology 2008, 9:R111 (doi:10.1186/gb-2008-9-7-r111) Accepted: 9 July 2008 The electronic version of this article is the complete one and can be found online at http://genomebiology.com/2008/9/7/R111 © 2008 Lukas et al.; licensee BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Epidemiological and genetic studies indicate that ethnic/genetic background plays an important role in susceptibility to primary open angle glaucoma (POAG). -

Global Analysis of Trna and Translation Factor Expression Reveals a Dynamic Landscape of Translational Regulation in Human Cancers
ARTICLE https://doi.org/10.1038/s42003-018-0239-8 OPEN Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers Zhao Zhang1, Youqiong Ye1, Jing Gong1, Hang Ruan1, Chun-Jie Liu2, Yu Xiang1, Chunyan Cai3, An-Yuan Guo2, 1234567890():,; Jiqiang Ling4, Lixia Diao5, John N. Weinstein5 & Leng Han 1,6 The protein translational system, including transfer RNAs (tRNAs) and several categories of enzymes, plays a key role in regulating cell proliferation. Translation dysregulation also contributes to cancer development, though relatively little is known about the changes that occur to the translational system in cancer. Here, we present global analyses of tRNAs and three categories of enzymes involved in translational regulation in ~10,000 cancer patients across 31 cancer types from The Cancer Genome Atlas. By analyzing the expression levels of tRNAs at the gene, codon, and amino acid levels, we identified unequal alterations in tRNA expression, likely due to the uneven distribution of tRNAs decoding different codons. We find that overexpression of tRNAs recognizing codons with a low observed-over-expected ratio may overcome the translational bottleneck in tumorigenesis. We further observed overall overexpression and amplification of tRNA modification enzymes, aminoacyl-tRNA synthe- tases, and translation factors, which may play synergistic roles with overexpression of tRNAs to activate the translational systems across multiple cancer types. 1 Department of Biochemistry and Molecular Biology, McGovern Medical School at The University of Texas Health Science Center at Houston, Houston, TX 77030, USA. 2 Department of Bioinformatics and Systems Biology, Hubei Bioinformatics and Molecular Imaging Key Laboratory, Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life Science and Technology, Huazhong University of Science and Technology Wuhan, 430074 Hubei, People’s Republic of China. -

Discovery, Expression Profiling, and Evolutionary Analysis Of
DISCOVERY, EXPRESSION PROFILING, AND EVOLUTIONARY ANALYSIS OF CYNODON EXPRESSED SEQUENCE TAGS by CHANGSOO KIM (Under the Direction of Andrew H. Paterson) ABSTRACT Bermudagrass (Cynodon dactylon) is a major turfgrass species for sports fields, lawns, parks, golf courses, and general utility turfs in tropical and subtropical regions. Despite its ecological importance, much of its study has been dependent upon classical approaches. Information about Bermudagrass at the molecular level has been deficient although molecular information for other plants has been accumulated for the last two dacades. In the current study, we constructed a normalized cDNA library from leaf tissue of Bermudagrass in order to expand our knowledge of its transcriptome. We sequenced and annotated 15,588 expressed sequence tags (ESTs), which were deposited in the National Center for Biotechnology Information (NCBI) to be shared with other scientists. We also conducted cDNA array hybridization (macroarray) to profile genes responding to drought stress. A total of 120 and 69 genes were identified as up- and down-regulated, respectively. BLASTX annotation suggested that up-regulated genes may be involved in osmotic adjustment, signal transduction pathways, protein repair systems, and removal of toxins, while down-regulated genes were mostly related to basic plant metabolism such as photosynthesis and glycolysis. Using the cDNA sequences, we performed a comparative genomic study to gain new insight into the evolution of Bermudagrass. Results suggested that the common ancestor of the grass family experienced a whole genome duplication event at ca. 50.0 ~ 65.4 million years ago (MYA), before the divergence of the PACC and BEP clades at ca. 42.3 ~ 50.0 MYA. -

Platform Abstracts
American Society of Human Genetics 65th Annual Meeting October 6–10, 2015 Baltimore, MD PLATFORM ABSTRACTS Wednesday, October 7, 9:50-10:30am Abstract #’s Friday, October 9, 2:15-4:15 pm: Concurrent Platform Session D: 4. Featured Plenary Abstract Session I Hall F #1-#2 46. Hen’s Teeth? Rare Variants and Common Disease Ballroom I #195-#202 Wednesday, October 7, 2:30-4:30pm Concurrent Platform Session A: 47. The Zen of Gene and Variant 15. Update on Breast and Prostate Assessment Ballroom III #203-#210 Cancer Genetics Ballroom I #3-#10 48. New Genes and Mechanisms in 16. Switching on to Regulatory Variation Ballroom III #11-#18 Developmental Disorders and 17. Shedding Light into the Dark: From Intellectual Disabilities Room 307 #211-#218 Lung Disease to Autoimmune Disease Room 307 #19-#26 49. Statistical Genetics: Networks, 18. Addressing the Difficult Regions of Pathways, and Expression Room 309 #219-#226 the Genome Room 309 #27-#34 50. Going Platinum: Building a Better 19. Statistical Genetics: Complex Genome Room 316 #227-#234 Phenotypes, Complex Solutions Room 316 #35-#42 51. Cancer Genetic Mechanisms Room 318/321 #235-#242 20. Think Globally, Act Locally: Copy 52. Target Practice: Therapy for Genetic Hilton Hotel Number Variation Room 318/321 #43-#50 Diseases Ballroom 1 #243-#250 21. Recent Advances in the Genetic Basis 53. The Real World: Translating Hilton Hotel of Neuromuscular and Other Hilton Hotel Sequencing into the Clinic Ballroom 4 #251-#258 Neurodegenerative Phenotypes Ballroom 1 #51-#58 22. Neuropsychiatric Diseases of Hilton Hotel Friday, October 9, 4:30-6:30pm Concurrent Platform Session E: Childhood Ballroom 4 #59-#66 54. -

Variation in Protein Coding Genes Identifies Information Flow
bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 1 1 2 3 4 5 Variation in protein coding genes identifies information flow as a contributor to 6 animal complexity 7 8 Jack Dean, Daniela Lopes Cardoso and Colin Sharpe* 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Institute of Biological and Biomedical Sciences 25 School of Biological Science 26 University of Portsmouth, 27 Portsmouth, UK 28 PO16 7YH 29 30 * Author for correspondence 31 [email protected] 32 33 Orcid numbers: 34 DLC: 0000-0003-2683-1745 35 CS: 0000-0002-5022-0840 36 37 38 39 40 41 42 43 44 45 46 47 48 49 Abstract bioRxiv preprint doi: https://doi.org/10.1101/679456; this version posted June 21, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Animal complexity and information flow 2 1 Across the metazoans there is a trend towards greater organismal complexity. How 2 complexity is generated, however, is uncertain. Since C.elegans and humans have 3 approximately the same number of genes, the explanation will depend on how genes are 4 used, rather than their absolute number. -

(B6;129.Cg-Gt(ROSA)26Sor Tm20(CAG-Ctgf-GFP)Jsd) Were Crossed with Female Foxd1cre/+ Heterozygote Mice 1, and Experimental Mice Were Selected As Foxd1cre/+; Rs26cig/+
Supplemental Information SI Methods Animal studies Heterozygote mice (B6;129.Cg-Gt(ROSA)26Sor tm20(CAG-Ctgf-GFP)Jsd) were crossed with female Foxd1Cre/+ heterozygote mice 1, and experimental mice were selected as Foxd1Cre/+; Rs26CIG/+. In some studies Coll-GFPTg or TCF/Lef:H2B-GFPTg mice or Foxd1Cre/+; Rs26tdTomatoR/+ mice were used as described 2; 3. Left kidneys were subjected to ureteral obstruction using a posterior surgical approach as described 2. In some experiments recombinant mouse DKK1 (0.5mg/kg) or an equal volume of vehicle was administered by daily IP injection. In the in vivo ASO experiment, either specific Lrp6 (TACCTCAATGCGATTT) or scrambled negative control ASO (AACACGTCTATACGC) (30mg/kg) (Exiqon, LNA gapmers) was administered by IP injection on d-1, d1, d4, and d7. In other experiments anti-CTGF domain-IV antibodies (5mg/kg) or control IgG were administered d-1, d1 and d6. All animal experiments were performed under approved IACUC protocols held at the University of Washington and Biogen. Recombinant protein and antibody generation and characterization Human CTGF domain I (sequence Met1 CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFC), domain I+II (sequence Met1CPDEPAPRCPAGVSLVLDGCGCCRVCAKQLGELCTERDPCDPHKGLFCCIFGGT VYRSGESFQSSCKYQCTCLDGAVGCMPLCSMDVRLPSPDCPFPRRVKLPGKCCEE) were cloned and expressed in 293 cells, and purified by Chelating SFF(Ni) Column, tested for single band by SEC and PAGE, and tested for absence of contamination. Domain-IV (sequence GKKCIRTPKISKPIKFELSGCTSMKTYRAKFCGVCTDGRCCTPHRTTTLPVEFKCPDGE VMKKNMMFIKTCACHYNCPGDNDIFESLYYRKMY) was purchased from Peprotech. Mouse or human DKK1 was generated from the coding sequence with some modifications and a tag. Secreted protein was harvested from 293 cells, and purified by nickel column, and tested for activity in a supertopflash (STF) assay 4. DKK1 showed EC50 of 0.69nM for WNT3a-induced WNT signaling in STF cells. -

Bioinformatics Analysis for Multiple Gene Expression Profiles in Sepsis
DATABASE ANALYSIS e-ISSN 1643-3750 © Med Sci Monit, 2020; 26: e920818 DOI: 10.12659/MSM.920818 Received: 2019.10.19 Accepted: 2020.01.21 Bioinformatics Analysis for Multiple Gene Available online: 2020.02.17 Published: 2020.04.13 Expression Profiles in Sepsis Authors’ Contribution: B Jianhua Zhai Department of Emergency, Tianjin Medical University General Hospital, Tianjin, Study Design A C Anlong Qi P.R. China Data Collection B Statistical Analysis C D Yan Zhang Data Interpretation D E Lina Jiao Manuscript Preparation E F Yancun Liu Literature Search F Funds Collection G A Songtao Shou Corresponding Author: Songtao Shou, e-mail: [email protected] Source of support: This work was supported by the National Natural Science Foundation of China (Grant No. 81871593) Background: This work aimed to screen key biomarkers related to sepsis progression by bioinformatics analyses. Material/Methods: The microarray datasets of blood and neutrophils from patients with sepsis or septic shock were download- ed from Gene Expression Omnibus database. Then, differentially expressed genes (DEGs) from 4 groups (sep- sis versus normal blood samples; septic shock versus normal blood samples; sepsis neutrophils versus normal controls and septic shock neutrophils versus controls) were respectively identified followed by functional anal- yses. Subsequently, protein–protein network was constructed, and key functional sub-modules were extracted. Finally, receiver operating characteristic analysis was conducted to evaluate diagnostic values of key genes. Results: There were 2082 DEGs between blood samples of sepsis patients and controls, 2079 DEGs between blood sam- ples of septic shock patients and healthy individuals, 6590 DEGs between neutrophils from sepsis and con- trols, and 1056 DEGs between neutrophils from septic shock patients and normal controls. -
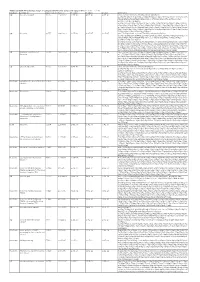
Additional Tables.Xlsx
Additional Table 5 Enriched pathways of upregulated DEGs after spinal cord injury in WT (P < 0.05, q < 0.05) Database Description Ratio of DEGs Ratio of P -value P adjust q value gene name GO ribosomal subunit 87/755 214/12262 1.03E-49 4.61E-47 4.20E-47 Gm6576/Gm10036/Gm5786/Mrps28/Gm9493/Rpl13- ps3/Rpl7l1/Mrpl10/Mrpl44/Rps20/Mrpl51/Mrps11/Mpv17l2/Mrps7/Gm10020/Gm10073 /Mrpl2/Mrpl52/Rpl22/Mrpl4/Mrps24/Zcchc17/Rpl28/Mrpl34/Rpl29/Mrps36/Rpl9- ps6/Hba-a2/Hba-a1/Rpl23a- ps3/Rpl15/Rack1/Mrps18a/Mrpl28/Rpl10a/Mrpl12/Rpl18/Rpl35a/Mrpl41/Mrps12/Rpl36 a/Rps9/Rps18/Rps5/Rps27a/Rps24/Rplp2/Rpl14/Mrpl30/Rps6/Rpl6/Rps3/Rps4x/Rpsa/R pl7/Rpl27a/Rps23/Rps10/Mrps21/Rpl36/Rpl30/Rps17/Mrpl42/Rpl35/Rpl27/Rplp0/Rps28 /Rpl31/Rpl39/Rps13/Rpl13/Rps11/Rpl8/Rps3a1/Fau/Rpl9/Rpl3/Rpl26/Rps15/Rpl13a/Uba 52/Rpl24/Rpl19/Rps14/Rpl4/Rps27/Rplp1 GO ribosome 93/755 248/12262 1.67E-49 4.61E-47 4.20E-47 Gm6576/Pnpt1/Gm10036/Gm5786/Mrps28/Gm9493/Rpl13- ps3/Rpl7l1/Mrpl10/Mrpl44/Rps20/Mrpl51/Mrps11/Mpv17l2/Mrps7/Gm10020/Gm10073 /Mrpl2/Mrpl52/Rpl22/Mrpl4/Mrps24/Zcchc17/Rpl28/Mrpl34/Rpl29/Mrps36/Rpl9- ps6/Hba-a2/Hba-a1/Rpl23a- ps3/Rpl15/Rack1/Eif3h/Mrps18a/Mrpl28/Mrps33/Rpl10a/Mrpl12/Rpl18/Rpl35a/Btf3/Mrp l41/Mrps12/Rpl36a/Rps9/Rps18/Rps5/Rps27a/Rps24/Rplp2/Rpl14/Mrpl30/Rps6/Rpl6/R ps3/Rps4x/Rpsa/Rpl7/Rpl27a/Rps23/Rps10/Mrps21/Rpl36/Rpl30/Rps17/Mrpl42/Rpl35/R pl27/Rplp0/Rps28/Rpl31/Rpl39/Rps13/Rpl13/Rps11/Rpl8/Rps3a1/Fau/Rpl9/Rpl3/Rpl26/ Rps15/Rpl13a/Uba52/Rpl41/Rpl24/Rpl19/Rps14/Rpl4/Ndufa7/Rps27/Rplp1 GO structural constituent of 76/733 169/11958 1.65E-47 1.16E-44 1.12E-44 Gm6576/Gm10036/Gm5786/Rpl7l1/Mrpl10/Rps20/Mrpl51/Mrps11/Mrps7/Gm10020/G