Magnesium Hydroxide Sorbents for Combined Carbon Dioxide Capture and Storage in Energy Conversion Systems
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Purified Sea Salt with Magnesium Carbonate
Cargill® Food Processing Salts Purified Sea Saltwith Magnesium Carbonate Product Description Physical Information Purified Sea Salt with Magnesium Carbonate This material is a food grade, granular, white crystalline Purified Sea Salt with Magnesium Carbonate sodium chloride product manufactured under stringent PHYSICAL MIN TARGET MAX process control procedures. Cargill Sea Salts are made from Pacific Ocean sea salt, which is harvested from ponds NaCl (%) 99.7 99.96 100 near the San Francisco Bay. Ca & Mg as Ca (%) 0.003 Sulfate as SO4 (%) 0.01 Product Application Water Insolubles (%) 0.025 0.01 Bulk Density (#cu/ft) 69 74 84 This material is intended for table and cooking use, as well as direct application in foods manufactured by the various Bulk Density (g/l) 1105 1185 1345 food processing industries. This material contains Surface Moisture (%) 0.02 Magnesium Carbonate, which is added to improve caking Magnesium Carbonate (%) 0.5 resistance and flowability. PERCENT PARTICLE SIZE MIN TARGET MAX Product Certifications DISTRIBUTION (SCREENS) Sieve - USS 30 Mesh Retained 0 40 50 Cargill® Sea Salts meet USDA, FDA and Food Chemicals Codex for food use. Sieve - USS 40 Mesh Retained 34 Sieve - USS 50 Mesh Retained 16 Cargill® Sea Salts are certified Kosher for Passover (OU-P) SieveCargill - USS 70 Mesh® RetainedSea Salt 8 by the Orthodox Union. Sieve - Retained on Pan 0 1 10 Made with Sun, Wind and Time Allergen Status Harvesting sea salt from San Francisco Bay today is similar to the salt-making process that has been used for centuries. In accordance with the 2004 USA Food Allergen Labeling and Consumer Protection Act (FALCPA), no allergen declarations are required for this product. -

Medication Other Information Aches and Pains Constipation Cough/Cold Diarrhea Fever Over the Counter Medications in Pregnancy
Over the Counter Medications in Pregnancy Women commonly use over the counter medications in pregnancy. This is a list of those medication you may safely use during pregnancy. If you have any questions about these medications and how to use them please contact your Healthcare Provider's office. Unless otherwise stated please take the medication as directed on the manufactures label. Medication Other Information Aches and Pains * Tylenol Extra Strength 500 mg tablet No more than 6 tablets in a 24 hour period * Tylenol 325 mg tablet No more than 8 in a 24 hour period Constipation Stool Softener Increases the amount of water in your stools to make them easier to pass * Colace (docusate sodium) * Surfak (docusate calcium) * Docusate Fiber Laxative Increases the amount of bulk in your stools to make them easier to pass * Metamucil (psyllium) * Fibercon (Calcium polycarbophil 625 mg) Stool Softener/Fiber Laxative Increases the amount of water in your stools to make them easier to pass * Peri-Colace (docusate sodium/sennosides) * Senekot -S (docusate sodium/sennosides) Osmotic Laxative Increases the amount of water in your stools to make them easier to pass * Milk of Magnesia * MiraLax (polyethylene glycol 3350) Cough/Cold Expectorants Help thin mucus and phlegm so they can be coughed up * Robitussin * Guaifenesin Antihistamines Can be used to relieve seasonal allergy and common cold symptoms of nasal congestion, * Chlorpheniramine sneezing and itchy eyes Cough Suppressants Help calm a cough * Dextromethorphan Decongestants Are used to relieve -

United States Patent Office Patented Nov
3,539,306 United States Patent Office Patented Nov. 10, 1970 2 properties of quick activity, durable effect and storage 3,539,306 PROCESS FOR THE PREPARATION Stability, and that, therefore, the same can provide an ex OF HYDROTALCITE cellent, ideal antacid. Teruhiko Kumura and Norio Imataki, Takamatsu-shi, Hydrotalcite is known as a mineral having a chemical Katuyuki Hasui, Kagawa-ken, and Takeo Inoue and 5 Structure of the formula Kimiaki Yasutomi, Nagao-machi, Kagawa-ken, Japan, assignors to Kyowa Chemical Industry Co., Ltd., Tokyo, MgAl(OH) 16CO3 4HO O Al2O3 6MgO CO2: 12H2O Japan, a corporation of Japan No Drawing. Filed July 21, 1967, Ser. No. 654,977 which has been naturally produced in only very limited Claims priority, application Japan, July 25, 1966, areas as Norway and Ural. Also a process for the syn 41/48,349; July 17, 1967, 42/45,658 O thesis of hydrotalcite was disclosed, which comprises add Int. C. COf 5/00, 7/00, A61k 27/06 ing Dry Ice or ammonium carbonate to a mixture of U.S. C. 23-315 4 Claims magnesium oxide and y-alumina or the thermal decom position product from of magnesium nitrate and aluminum nitrate, and thereafter maintaining the system at tempera ABSTRACT OF THE DISCLOSURE, tures below 325 C. and under elevated pressures of total A process for the preparation of hydrotalcite which 2,000-20,000 p.s.i. (Roy et al.: American Journal of comprises mixing an alumium component such as alumi Science 251, 350-353 (1953)). However this process is num Salt With a magnesium component such as magnesium obviously unpractical for industrial scale production of Salt in an aqueous medium in the presence of carbonate hydrotalcite, because the reaction system must be main ion at a pH of at least 8 and thereafter recovering the 20 tained under considerably high pressures. -

Disposal of Solid Chemicals in the Normal Trash
Disposal of Solid Chemicals in the Normal Trash Many solid chemicals can be safety discarded into the normal trash, provided they are in containers that are not broken or cracked and have tightly fitting caps. These chemicals are considered acceptable for ordinary disposal because they display none of the properties of hazardous waste, are of low acute toxicity, and have not been identified as having any chronic toxic effects as summarized in the National Institute of Occupational Safety and Health (NIOSH) “Registry of Toxic Effects of Chemical Substances”. Examples of chemicals acceptable for disposal as regular trash are listed below. To dispose of these chemicals, place the containers in a box lined with a plastic bag, tape the top of the box shut, write “Normal Trash” on the box and then place the box next to the lab trash container. Only solid forms of these chemicals can be disposed in this manner. Any questions about these chemicals or other chemicals that may be disposed of in the normal trash should be directed to the Hazardous Materials Technician (610) 330-5225. Chemicals Generally Acceptable for Disposal as Regular Trash Acacia powder, gum Detergent (most) Methyl salicylate Sodium carbonate arabic Cation exchange resins Methylene blue Sodium chloride Acid, Ascorbic Chromatographic Methyl stearate Sodium citrate Acid, Benzoic absorbents Nutrient agar Sodium dodecyl sulfate Acid, Boric Crystal violet Octacosane (SDS) Acid, Casamind Dextrin Parafin Sodium formate Acid, Citric Dextrose Pepsin Sodium iodide Acid, Lactic Diatomaceous -

Dietary Supplements Compendium Volume 1
2015 Dietary Supplements Compendium DSC Volume 1 General Notices and Requirements USP–NF General Chapters USP–NF Dietary Supplement Monographs USP–NF Excipient Monographs FCC General Provisions FCC Monographs FCC Identity Standards FCC Appendices Reagents, Indicators, and Solutions Reference Tables DSC217M_DSCVol1_Title_2015-01_V3.indd 1 2/2/15 12:18 PM 2 Notice and Warning Concerning U.S. Patent or Trademark Rights The inclusion in the USP Dietary Supplements Compendium of a monograph on any dietary supplement in respect to which patent or trademark rights may exist shall not be deemed, and is not intended as, a grant of, or authority to exercise, any right or privilege protected by such patent or trademark. All such rights and privileges are vested in the patent or trademark owner, and no other person may exercise the same without express permission, authority, or license secured from such patent or trademark owner. Concerning Use of the USP Dietary Supplements Compendium Attention is called to the fact that USP Dietary Supplements Compendium text is fully copyrighted. Authors and others wishing to use portions of the text should request permission to do so from the Legal Department of the United States Pharmacopeial Convention. Copyright © 2015 The United States Pharmacopeial Convention ISBN: 978-1-936424-41-2 12601 Twinbrook Parkway, Rockville, MD 20852 All rights reserved. DSC Contents iii Contents USP Dietary Supplements Compendium Volume 1 Volume 2 Members . v. Preface . v Mission and Preface . 1 Dietary Supplements Admission Evaluations . 1. General Notices and Requirements . 9 USP Dietary Supplement Verification Program . .205 USP–NF General Chapters . 25 Dietary Supplements Regulatory USP–NF Dietary Supplement Monographs . -

Medications and the Gluten-Free Diet
Resources for GF Drug Medications and the Information: Gluten-Free Diet - www.glutenfreedrugs.com (Pharmacist controlled site) Updated January 2021 - dailymed.nlm.nih.gov/ Staying gluten-free involves more than just dietary changes.While use of dailymed/ gluten as an ingredient in medications is uncommon, medications must also be evaluated for their gluten content. Many pharmaceuticals have an additional filler called an excipient, used to make a particular dosage form of the drug. These fillers are often made from corn, potato, or tapioca. However, some are derived This educational bulletin has been from wheat or, rarely, barley. If you have doubts about the gluten-free status of a produced by the Gluten Intolerance medication, it is important that you consult with your pharmacist and/or check with Group of North America, a registered the manufacturer of the product. 501(c)3 organization. Learn more about GIG at www.gluten.org. The majority of drug manufacturers do not clearly label their products or packaging with information regarding the gluten content of their drug. Medications GIG is on a mission to make life easier – whether prescription or over-the-counter - are not included in the FDA gluten- for everyone living gluten-free. free labeling regulation which applies to FDA-regulated food products. In late 2017 the FDA issued a draft document on labeling recommendations for medications; as of May 2018 the guidelines were not finalized. If and when This information is for educational purposes only. Consult your such guidelines are finalized, consumers may have easier access to information healthcare team when considering about the gluten content of medications. -

Estonian Statistics on Medicines 2016 1/41
Estonian Statistics on Medicines 2016 ATC code ATC group / Active substance (rout of admin.) Quantity sold Unit DDD Unit DDD/1000/ day A ALIMENTARY TRACT AND METABOLISM 167,8985 A01 STOMATOLOGICAL PREPARATIONS 0,0738 A01A STOMATOLOGICAL PREPARATIONS 0,0738 A01AB Antiinfectives and antiseptics for local oral treatment 0,0738 A01AB09 Miconazole (O) 7088 g 0,2 g 0,0738 A01AB12 Hexetidine (O) 1951200 ml A01AB81 Neomycin+ Benzocaine (dental) 30200 pieces A01AB82 Demeclocycline+ Triamcinolone (dental) 680 g A01AC Corticosteroids for local oral treatment A01AC81 Dexamethasone+ Thymol (dental) 3094 ml A01AD Other agents for local oral treatment A01AD80 Lidocaine+ Cetylpyridinium chloride (gingival) 227150 g A01AD81 Lidocaine+ Cetrimide (O) 30900 g A01AD82 Choline salicylate (O) 864720 pieces A01AD83 Lidocaine+ Chamomille extract (O) 370080 g A01AD90 Lidocaine+ Paraformaldehyde (dental) 405 g A02 DRUGS FOR ACID RELATED DISORDERS 47,1312 A02A ANTACIDS 1,0133 Combinations and complexes of aluminium, calcium and A02AD 1,0133 magnesium compounds A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 811120 pieces 10 pieces 0,1689 A02AD81 Aluminium hydroxide+ Magnesium hydroxide (O) 3101974 ml 50 ml 0,1292 A02AD83 Calcium carbonate+ Magnesium carbonate (O) 3434232 pieces 10 pieces 0,7152 DRUGS FOR PEPTIC ULCER AND GASTRO- A02B 46,1179 OESOPHAGEAL REFLUX DISEASE (GORD) A02BA H2-receptor antagonists 2,3855 A02BA02 Ranitidine (O) 340327,5 g 0,3 g 2,3624 A02BA02 Ranitidine (P) 3318,25 g 0,3 g 0,0230 A02BC Proton pump inhibitors 43,7324 A02BC01 Omeprazole -

DIGESTIF RENNIE ANISEED Tablets Should Be Used with Caution When a State of Debility, Renal Insufficiency Or a Pre-Disposition to Kidney Stones Exists
PROFESSIONAL INFORMATION SCHEDULING STATUS: S0 PROPRIETARY NAME: COMPOSITION: Each tablet contains: Active ingredients: Calcium carbonate 680 mg and magnesium carbonate 80 mg. Inactive ingredients: Aniseed flavour, aspartame*, light liquid paraffin, liquorice flavour, magnesium stearate, maize starch, potato starch, sucrose, talc. Contains sugar: sucrose 475 mg/ tablet. Contains sweetener: aspartame 10 mg/tablet *Phenylketonurics: contains a source phenylalanine. CATEGORY AND CLASS: A / 11.4.1 - Acid Neutraliser PHARMACOLOGICAL ACTION: Antacid INDICATIONS: When an antacid is indicated, such as indigestion and heartburn. CONTRA-INDICATIONS: Hypermagnesaemia, Hypercalcaemia or alkalosis. Hypersensitivity to any of the ingredients. Page 1 of 4 PROFESSIONAL INFORMATION WARNINGS AND SPECIAL PRECAUTIONS: Do not continue usage for longer than 2 weeks except on the advice of a doctor. Do not give to children under five years, except on the advice of a doctor. May interfere with the adsorption of other medicines given concomitantly. DOSAGE AND DIRECTIONS FOR USE: Adults and children ≥ 12 years: 1-2 chewable tablets as a single dose, preferably to be taken one hour after meals and before going to bed but also between meals in case of heartburn or epigastric pain. A maximum daily dose of 8 g calcium carbonate (corresponding to 11 Digestif Rennie® tablets) must not be exceeded. If symptoms persist in spite of therapy, diagnostic measures are strongly recommended in order to rule out a more serious disease. Not recommended for children under 12 years of age. Method of administration: For oral use. The tablets are to be chewed or sucked. No water is required. SIDE EFFECTS: Calcium carbonate may cause constipation. However, the Magnesium carbonate may affect the latter tendency since magnesium salts are known to be mildly laxative in action. -

Solid Chemicals for the Normal Trash
SOLID CHEMICALS FOR THE NORMAL TRASH Solid chemicals acceptable for disposal as regular trash are listed below: Acacia powder, gum arabic Calcium silicate Manganese sulfate Acid, Ascorbic Calcium sulfate Methyl red Acid, Benzoic Detergent (most) Methyl salicylate Acid, Boric Cation exchange resins Methylene blue Acid, Casamind Crystal violet Methyl stearate Acid, Citric Dextrin Nutrient agar Acid, Lactic Dextrose Octacosane Acid, Oleic Diatomaceous earth Parafin Acid, Phthalic Docosanoic acid Pepsin Acid, Salicycle Drierite (calcium sulfate, anhydrous) Peptone Acid, Silicic Ferric oxide Petroleum jelly Acid, Stearic Ferric phosphate Polyethylene, solid Acid, Succinic Ferric pyrophosphate Polystryene Acid, Tartaric Ferric sulfate Potassium acetate Acrylamide gels Ferrous ammonium sulfate Potassium bicarbonate Agar(s) Galactose Potassium bromide Albumen Geletin Potassium carbonate Alumina Gum arabic Potassium chloride Aluminum oxide Gum guaiac Potassium citrate Amino acids, naturally occurring Hexadecanol, 1- Potassium ferricyanide Ammonium bicarbonate Kaolin Potassium iodide Ammonium phosphate Lactose Potassium phosphate Ammonium sulfate Lanolin Potassium sodium tartrate Ammonium sulfamate Lauric acid Potassium sulfate Base, blood agar Lauryl sulfate Potassium sulfite Beef extract Lithium carbonate Potassium sulfocyanate Behenic acid Lithium chloride Pumice Bentonite Lithium sulfate Salts, naturally occurring Brain heart infusion Litmus Sand Bromphenol blue Magnesium carbonate Silica Broth, nutrient Magnesium chloride Silica gel, unused -

EUROPEAN PHARMACOPOEIA 10.0 Index 1. General Notices
EUROPEAN PHARMACOPOEIA 10.0 Index 1. General notices......................................................................... 3 2.2.66. Detection and measurement of radioactivity........... 119 2.1. Apparatus ............................................................................. 15 2.2.7. Optical rotation................................................................ 26 2.1.1. Droppers ........................................................................... 15 2.2.8. Viscosity ............................................................................ 27 2.1.2. Comparative table of porosity of sintered-glass filters.. 15 2.2.9. Capillary viscometer method ......................................... 27 2.1.3. Ultraviolet ray lamps for analytical purposes............... 15 2.3. Identification...................................................................... 129 2.1.4. Sieves ................................................................................. 16 2.3.1. Identification reactions of ions and functional 2.1.5. Tubes for comparative tests ............................................ 17 groups ...................................................................................... 129 2.1.6. Gas detector tubes............................................................ 17 2.3.2. Identification of fatty oils by thin-layer 2.2. Physical and physico-chemical methods.......................... 21 chromatography...................................................................... 132 2.2.1. Clarity and degree of opalescence of -

Syntheses of Nanostructured Magnesium Carbonate Powders with Mesoporous Structures from Carbon Dioxide
applied sciences Article Syntheses of Nanostructured Magnesium Carbonate Powders with Mesoporous Structures from Carbon Dioxide Fernando J. Rodríguez-Macías 1,2 , José E. Ortiz-Castillo 1 , Erika López-Lara 1, Alejandro J. García-Cuéllar 1 , José L. López-Salinas 1 ,César A. García-Pérez 1, Orlando Castilleja-Escobedo 1 and Yadira I. Vega-Cantú 1,2,* 1 Tecnologico de Monterrey, Escuela de Ingeniería y Ciencias, Ave. Eugenio Garza Sada 2501, Monterrey 64849, N.L., Mexico; [email protected] (F.J.R.-M.); [email protected] (J.E.O.-C.); [email protected] (E.L.-L.); [email protected] (A.J.G.-C.); [email protected] (J.L.L.-S.); [email protected] (C.A.G.-P.); [email protected] (O.C.-E.) 2 Pós-Graduação em Ciência de Materiais, Universidade Federal de Pernambuco, Av. Prof. Moraes Rego 1235, Cidade Universitária, Recife 50.670-901, PE, Brazil * Correspondence: [email protected] Abstract: In this work, we present the results of two synthesis approaches for mesoporous magnesium carbonates, that result in mineralization of carbon dioxide, producing carbonate materials without the use of cosolvents, which makes them more environmentally friendly. In one of our synthesis methods, we found that we could obtain nonequilibrium crystal structures, with acicular crystals branching bidirectionally from a denser core. Both Raman spectroscopy and X-ray diffraction showed these crystals to be a mixture of sulfate and hydrated carbonates. We attribute the nonequilibrium morphology to coprecipitation of two salts and short synthesis time (25 min). Other aqueous synthesis conditions produced mixtures of carbonates with different morphologies, which changed depending on drying temperature (40 or 100 ◦C). -
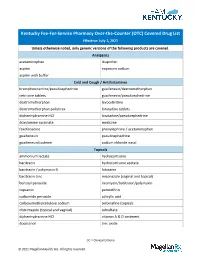
OTC) Covered Drug List Effective: July 1, 2021 Unless Otherwise Noted, Only Generic Versions of the Following Products Are Covered
Kentucky Fee-For-Service Pharmacy Over-the-Counter (OTC) Covered Drug List Effective: July 1, 2021 Unless otherwise noted, only generic versions of the following products are covered. Analgesics acetaminophen ibuprofen aspirin naproxen sodium aspirin with buffer Cold and Cough / Antihistamines brompheniramine/pseudoephedrine guaifenesin/dextromethorphan cetirizine tablets guaifenesin/pseudoephedrine dextromethorphan levocetirizine dextromethorphan polistirex loratadine tablets diphenhydramine HCl loratadine/pseudoephedrine doxylamine succinate meclizine fexofenadine phenylephrine / acetaminophen guaifenesin pseudoephedrine guaifenesin/codeine sodium chloride nasal Topicals ammonium lactate hydrocortisone bacitracin hydrocortisone acetate bacitracin / polymyxin B lidocaine bacitracin zinc miconazole (vaginal and topical) benzoyl peroxide neomycin/bacitracin/polymyxin capsaicin permethrin carbamide peroxide salicylic acid carboxymethylcellulose sodium terbinafine (topical) clotrimazole (topical and vaginal) tolnaftate diphenhydramine HCl vitamin A & D ointment docosanol zinc oxide CC = Clinical Criteria © 2021 Magellan Health, Inc. All rights reserved. Vitamins ascorbic acid (Vitamin C) ferrous sulfate calcium acetate fish oil calcium carbonate / vitamin D3 folic acid calcium citrate magnesium oxide calcium citrate / vitamin D3 niacin /niacinamide cyanocobalamin (Vitamin B-12) prenatal vitamins CC ergocalciferol (vitamin D2) pyridoxine HCl (vitamin B6) ferrous gluconate vitamin D3 GI Products aluminum hydroxide magnesium carbonate / aluminum