The Synthesis of Anthranilic Acid, Tryptophan, and Sulfenyl Chloride Analogues, and Enzymatic Studies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
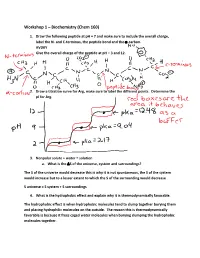
Workshop 1 – Biochemistry (Chem 160)
Workshop 1 – Biochemistry (Chem 160) 1. Draw the following peptide at pH = 7 and make sure to include the overall charge, label the N- and C-terminus, the peptide bond and the -carbon. AVDKY Give the overall charge of the peptide at pH = 3 and 12. 2. Draw a titration curve for Arg, make sure to label the different points. Determine the pI for Arg. 3. Nonpolar solute + water = solution a. What is the S of the universe, system and surroundings? The S of the universe would decrease this is why it is not spontaneous, the S of the system would increase but to a lesser extent to which the S of the surrounding would decrease S universe = S system + S surroundings 4. What is the hydrophobic effect and explain why it is thermodynamically favorable. The hydrophobic effect is when hydrophobic molecules tend to clump together burying them and placing hydrophilic molecules on the outside. The reason this is thermodynamically favorable is because it frees caged water molecules when burying clumping the hydrophobic molecules together. 5. Urea dissolves very readily in water, but the solution becomes very cold as the urea dissolves. How is this possible? Urea dissolves in water because when dissolving there is a net increase in entropy of the universe. The heat exchange, getting colder only reflects the enthalpy (H) component of the total energy change. The entropy change is high enough to offset the enthalpy component and to add up to an overall -G 6. A mutation that changes an alanine residue in the interior of a protein to valine is found to lead to a loss of activity. -

Free-Radical Chain Reactions of Organic Mercury and Tin Compounds Hasan I
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1984 Free-radical chain reactions of organic mercury and tin compounds Hasan I. Tashtoush Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Tashtoush, Hasan I., "Free-radical chain reactions of organic mercury and tin compounds " (1984). Retrospective Theses and Dissertations. 7733. https://lib.dr.iastate.edu/rtd/7733 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quality of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure complete continuity. 2. When an image on the film is obliterated with a round black mark, it is an indication of either blurred copy because of movement during exposure, duplicate copy, or copyrighted materials that should not have been filmed. -

THE CHEMISTRY of SULFENIMIDES by Barbara Ann Orwig a Thesis
THE CHEMISTRY OF SULFENIMIDES by Barbara Ann Orwig A thesis submitted to the Faculty of Graduate Studies and Research in partial fulfilment of the requirements for the degree of Master of Science Department of Chemistry McGill University Montreal, P.Q. Canada May 1971 @) Barbara Ann Orwig 1972 Dedicated to My Parents Il Thanx Il i ACKNOWLEDGEMENTS 3l MY thanks to Dr. D.F.R. Gilson for the p nmr spectra, to Victor Yu for the A-60 nmr spectra, and to Peter Currie for the mass spectra. For helpful discussions and worthwhile suggestions l thank David Ash 1 Errol Chang 1 and John Gleason. For his guidance and help as my research director l thank Dr. David N. Harpp. And to Elva and Hermann Heyge go special thanks for their help and for "putting up wi th œil. TABLE OF CONTENTS Page ACKNOWLEDGEMENTS i INTRODUcrION 1 EXPERIMENTAL SECTION 22 RESULTS AND DISCUSSION 42 TABLES 1 Preparation of Sulfenyl Chlorides 72 2 Preparation of N-(alkyl/aryl thio)phthalimides 73 3 Desulfurization Reactions of N-(alkyl/aryl thio)phthalimides 76 4 Mass Spectra of N-(alkyl/aryl thio)phthalimides 77 FIGURES (Spectra) 78 - 87 BIBLIOGRAPHY 88 INTRODUcrION AND BACKGROUND Sulfenic acids Cl), in which sulfur exists in its R-S-OH l lcwest oxidation state, are highly lmstable compolmds and only a few l have been isolated. The derivatives of sulfenic acid {~.> however, are generally isolable and usually stable. 2 R-S-Y 2 When Y is -NH ' -~HR, or -NR ' the resulting class of compolmds is 2 2 ter.med sulfenamides. -

On Glyphosate
Ecocycles 2(2): 1-8 (2016) ISSN 2416-2140 DOI: 10.19040/ecocycles.v2i2.60 EDITORIAL On glyphosate Tamas Komives1 * and Peter Schröder2 1Plant Protection Institute, Centre for Agricultural Research, Hungarian Academy of Sciences, Herman Otto 15, 1022 Budapest, Hungary and Department of Environmental Science, Esterhazy Karoly University, 3200 Gyongyos, Hungary 2Helmholtz Zentrum München, German Research Centre for Environmental Health, GmbH, Research Unit Environmental Genomics, Ingolstaedter Landstrasse 1, 85764 Neuherberg, Germany *E-mail: [email protected] Abstract – This Editorial briefly discusses the current issues surrounding glyphosate - the most controversial pesticide active ingredient of our time. The paper pays special attention to the effects of glyphosate on plant-pathogen interactions. Keywords – glyphosate, plant-pathogen interactions, environment, human health, ecocycles, sustainability Received: October 14, 2016 Accepted: November 10, 2016 ———————————————————————————————————————————————— - In nature nothing exists alone. researchers of the company missed to identify the Rachel Carson in “Silent Spring” (Carson, 1962) molecule as a potential herbicide because of the short duration of the company's standardized biological - Alle Dinge sind Gift, und nichts ist ohne Gift; allein assays (only five days, while the first, glyphosate- die Dosis machts, daß ein Ding kein Gift sei. (All induced phytotoxic symptoms usually appear after things are poison, and nothing is without poison: the about one week) (F. M. Pallos, -

APPENDIX G Acid Dissociation Constants
harxxxxx_App-G.qxd 3/8/10 1:34 PM Page AP11 APPENDIX G Acid Dissociation Constants § ϭ 0.1 M 0 ؍ (Ionic strength ( † ‡ † Name Structure* pKa Ka pKa ϫ Ϫ5 Acetic acid CH3CO2H 4.756 1.75 10 4.56 (ethanoic acid) N ϩ H3 ϫ Ϫ3 Alanine CHCH3 2.344 (CO2H) 4.53 10 2.33 ϫ Ϫ10 9.868 (NH3) 1.36 10 9.71 CO2H ϩ Ϫ5 Aminobenzene NH3 4.601 2.51 ϫ 10 4.64 (aniline) ϪO SNϩ Ϫ4 4-Aminobenzenesulfonic acid 3 H3 3.232 5.86 ϫ 10 3.01 (sulfanilic acid) ϩ NH3 ϫ Ϫ3 2-Aminobenzoic acid 2.08 (CO2H) 8.3 10 2.01 ϫ Ϫ5 (anthranilic acid) 4.96 (NH3) 1.10 10 4.78 CO2H ϩ 2-Aminoethanethiol HSCH2CH2NH3 —— 8.21 (SH) (2-mercaptoethylamine) —— 10.73 (NH3) ϩ ϫ Ϫ10 2-Aminoethanol HOCH2CH2NH3 9.498 3.18 10 9.52 (ethanolamine) O H ϫ Ϫ5 4.70 (NH3) (20°) 2.0 10 4.74 2-Aminophenol Ϫ 9.97 (OH) (20°) 1.05 ϫ 10 10 9.87 ϩ NH3 ϩ ϫ Ϫ10 Ammonia NH4 9.245 5.69 10 9.26 N ϩ H3 N ϩ H2 ϫ Ϫ2 1.823 (CO2H) 1.50 10 2.03 CHCH CH CH NHC ϫ Ϫ9 Arginine 2 2 2 8.991 (NH3) 1.02 10 9.00 NH —— (NH2) —— (12.1) CO2H 2 O Ϫ 2.24 5.8 ϫ 10 3 2.15 Ϫ Arsenic acid HO As OH 6.96 1.10 ϫ 10 7 6.65 Ϫ (hydrogen arsenate) (11.50) 3.2 ϫ 10 12 (11.18) OH ϫ Ϫ10 Arsenious acid As(OH)3 9.29 5.1 10 9.14 (hydrogen arsenite) N ϩ O H3 Asparagine CHCH2CNH2 —— —— 2.16 (CO2H) —— —— 8.73 (NH3) CO2H *Each acid is written in its protonated form. -

Amino Acid Chemistry
Handout 4 Amino Acid and Protein Chemistry ANSC 619 PHYSIOLOGICAL CHEMISTRY OF LIVESTOCK SPECIES Amino Acid Chemistry I. Chemistry of amino acids A. General amino acid structure + HN3- 1. All amino acids are carboxylic acids, i.e., they have a –COOH group at the #1 carbon. 2. All amino acids contain an amino group at the #2 carbon (may amino acids have a second amino group). 3. All amino acids are zwitterions – they contain both positive and negative charges at physiological pH. II. Essential and nonessential amino acids A. Nonessential amino acids: can make the carbon skeleton 1. From glycolysis. 2. From the TCA cycle. B. Nonessential if it can be made from an essential amino acid. 1. Amino acid "sparing". 2. May still be essential under some conditions. C. Essential amino acids 1. Branched chain amino acids (isoleucine, leucine and valine) 2. Lysine 3. Methionine 4. Phenyalanine 5. Threonine 6. Tryptophan 1 Handout 4 Amino Acid and Protein Chemistry D. Essential during rapid growth or for optimal health 1. Arginine 2. Histidine E. Nonessential amino acids 1. Alanine (from pyruvate) 2. Aspartate, asparagine (from oxaloacetate) 3. Cysteine (from serine and methionine) 4. Glutamate, glutamine (from α-ketoglutarate) 5. Glycine (from serine) 6. Proline (from glutamate) 7. Serine (from 3-phosphoglycerate) 8. Tyrosine (from phenylalanine) E. Nonessential and not required for protein synthesis 1. Hydroxyproline (made postranslationally from proline) 2. Hydroxylysine (made postranslationally from lysine) III. Acidic, basic, polar, and hydrophobic amino acids A. Acidic amino acids: amino acids that can donate a hydrogen ion (proton) and thereby decrease pH in an aqueous solution 1. -

The Use of Chlorosulfonic Acid in the Identification Of' Halogen Substitute):) Aromatic Compounds
THE USE OF CHLOROSULFONIC ACID IN THE IDENTIFICATION OF' HALOGEN SUBSTITUTE):) AROMATIC COMPOUNDS UI\LAt!UlU !GHirULTITRE & MECHANICAL COU"RI. ; LIBRA ]{Y i OCT 26 1937 THE USE OF CHLOROSULFONIC ACID IN THE IDENTIFICATION OF HALOGEN SUBSTITUTED AROMATIC COMPOUNDS By Elton Murray Baker,. Bachelor of Soience North.. estern State Teachers College 1955 Submitted to the Department of Chemistry Oklahoma Agricultural. and Mechanical. College In partial fulfillment of the requirements for the Degree of MASrER OF SCIENCE 1957 - ' ' • ~9 ! . • • • • r •: .. : : ... ..... .. ' .. .. ... ... .. .. ·.... ... .... ~ ~ ( . .. : : ( ~ · : ·. ... ' . • '' . OKLAHOMA ~liBICUL TURE &MECIL\NIO AL eniLEal ii LIBR ARY OCT 261937 APPROVED: In Charge of Thesis Head of the Department of Chemistry ~'~~Dean of the Graduate School. 100711 iii TABLE OF CONTENTS Page Introduction . • • • • • • • 1 Historical Review • • • 2 Experimental • • • • • • • • • 9 Discussion of Results • • .. • • • • 29 Summary • • • • • • • • • • • . 51 Bibliography . • • • ... • • • • • • 52 Autobiography • • • • • • • • • • • • • • • 56 iv ACKNOWLEDGEMENT The author wishes to express his sincere appreciation to Dr. o. c. Dermer under whose direction this work was done. He also .wishes to acknowledge the service rendered by t he Oklahoma Agricultural and Mechanical College Librarians and Chemistry Storeroom assistants. 1 INTBOOOO!'ION The identification of aromatic halides is generally accomplished by converting them into mono or pol.ynitro derivatives. The reaction is not always easy to control., however_. and sometimes gives mixtures hard to purify. In View of the rea<V' con'Vereion of aroma.tie hydrocarbons into solid s-ulfonyl chloride deri.Yatives by chloro.eulfonic acid~ it seemed profitable to extend the reaction to include aromatic halides. The method has the added advantage that, if a sulfonyl chloride is hard to plil'ify or does not uniquely identify a compound, it may he very ee{!ily converted into the suli'onamide,, which is general.ly a satisfactory derivative. -

Nucleotide Base Coding and Am1ino Acid Replacemients in Proteins* by Emil L
VOL. 48, 1962 BIOCHEMISTRY: E. L. SAIITH 677 18 Britten, R. J., and R. B. Roberts, Science, 131, 32 (1960). '9 Crestfield, A. M., K. C. Smith, and F. WV. Allen, J. Biol. Chem., 216, 185 (1955). 20 Gamow, G., Nature, 173, 318 (1954). 21 Brenner, S., these PROCEEDINGS, 43, 687 (1957). 22 Nirenberg, M. WV., J. H. Matthaei, and 0. WV. Jones, unpublished data. 23 Crick, F. H. C., L. Barnett, S. Brenner, and R. J. Watts-Tobin, Nature, 192, 1227 (1961). 24 Levene, P. A., and R. S. Tipson, J. Biol. Ch-nn., 111, 313 (1935). 25 Gierer, A., and K. W. Mundry, Nature, 182, 1437 (1958). 2' Tsugita, A., and H. Fraenkel-Conrat, J. Mllot. Biol., in press. 27 Tsugita, A., and H. Fraenkel-Conrat, personal communication. 28 Wittmann, H. G., Naturwissenschaften, 48, 729 (1961). 29 Freese, E., in Structure and Function of Genetic Elements, Brookhaven Symposia in Biology, no. 12 (1959), p. 63. NUCLEOTIDE BASE CODING AND AM1INO ACID REPLACEMIENTS IN PROTEINS* BY EMIL L. SMITHt LABORATORY FOR STUDY OF HEREDITARY AND METABOLIC DISORDERS AND THE DEPARTMENTS OF BIOLOGICAL CHEMISTRY AND MEDICINE, UNIVERSITY OF UTAH COLLEGE OF MEDICINE Communicated by Severo Ochoa, February 14, 1962 The problem of which bases of messenger or template RNA' specify the coding of amino acids in proteins has been largely elucidated by the use of synthetic polyri- bonucleotides.2-7 For these triplet nucleotide compositions (Table 1), it is of in- terest to examine some of the presently known cases of amino acid substitutions in polypeptides or proteins of known structure. -

Modulating Wine Aromatic Amino Acid Catabolites by Using Torulaspora Delbrueckii in Sequentially Inoculated Fermentations Or Saccharomyces Cerevisiae Alone
microorganisms Article Modulating Wine Aromatic Amino Acid Catabolites by Using Torulaspora delbrueckii in Sequentially Inoculated Fermentations or Saccharomyces cerevisiae Alone M. Antonia Álvarez-Fernández 1 , Ilaria Carafa 2, Urska Vrhovsek 2 and Panagiotis Arapitsas 2,* 1 Departamento de Nutrición y Bromatología, Toxicología y Medicina Legal, Facultad de Farmacia, Universidad de Sevilla, 41012 Sevilla, Spain; [email protected] 2 Department of Food Quality and Nutrition, Research and Innovation Centre, Fondazione Edmund Mach, 38010 San Michele all’Adige, Italy; [email protected] (I.C.); [email protected] (U.V.) * Correspondence: [email protected] or [email protected]; Tel.: +39-0461615656 Received: 25 August 2020; Accepted: 2 September 2020; Published: 4 September 2020 Abstract: Yeasts are the key microorganisms that transform grape juice into wine, and nitrogen is an essential nutrient able to affect yeast cell growth, fermentation kinetics and wine quality. In this work, we focused on the intra- and extracellular metabolomic changes of three aromatic amino acids (tryptophan, tyrosine, and phenylalanine) during alcoholic fermentation of two grape musts by two Saccharomyces cerevisiae strains and the sequential inoculation of Torulaspora delbrueckii with Saccharomyces cerevisiae. An UPLC-MS/MS method was used to monitor 33 metabolites, and 26 of them were detected in the extracellular samples and 8 were detected in the intracellular ones. The results indicate that the most intensive metabolomic changes occurred during the logarithm cellular growth phase and that pure S. cerevisiae fermentations produced higher amounts of N-acetyl derivatives of tryptophan and tyrosine and the off-odour molecule 2-aminoacetophenone. The sequentially inoculated fermentations showed a slower evolution and a higher production of metabolites linked to the well-known plant hormone indole acetic acid (auxin). -

The Role of Reduced Methionine in Mediating the Metabolic Responses to Protein Restriction Using Different Sources of Protein
nutrients Article The Role of Reduced Methionine in Mediating the Metabolic Responses to Protein Restriction Using Different Sources of Protein Han Fang 1 , Kirsten P. Stone 1 , Sujoy Ghosh 2,3 , Laura A. Forney 4 and Thomas W. Gettys 1,* 1 Laboratory of Nutrient Sensing & Adipocyte Signaling, 6400 Perkins Road, Pennington Biomedical Research Center, Baton Rouge, LA 70808, USA; [email protected] (H.F.); [email protected] (K.P.S.) 2 Laboratory of Computational Biology, Pennington Biomedical Research Center, Baton Rouge, LA 70808, USA; [email protected] 3 Program in Cardiovascular and Metabolic Disorders and Center for Computational Biology, Duke-NUS Medical School, Singapore 169857, Singapore 4 Department of Integrative Biology and Pharmacology, University of Texas Health Science Center at Houston, 7000 Fannin St, Houston, TX 77030, USA; [email protected] * Correspondence: [email protected] Abstract: Dietary protein restriction and dietary methionine restriction (MR) produce a comparable series of behavioral, physiological, biochemical, and transcriptional responses. Both dietary regimens produce a similar reduction in intake of sulfur amino acids (e.g., methionine and cystine), and both diets increase expression and release of hepatic FGF21. Given that FGF21 is an essential mediator of the metabolic phenotype produced by both diets, an important unresolved question is whether dietary protein restriction represents de facto methionine restriction. Using diets formulated from either casein or soy protein with matched reductions in sulfur amino acids, we compared the ability Citation: Fang, H.; Stone, K.P.; of the respective diets to recapitulate the metabolic phenotype produced by methionine restriction Ghosh, S.; Forney, L.A.; Gettys, T.W. -

Utilization of Anthranilic and Nitrobenzoic Acids by Nocardia Opaca and a Flavobacteriurn
J. gen. Microbial. (1966),42, 219-235 Printed in Great Britain Utilization of Anthranilic and Nitrobenzoic Acids by Nocardia opaca and a Flavobacteriurn BY R. B. CAIN Depadnaent of Microbiology, OkEahoma State University, Stillwater, Oklahoma, U.S.A. and Department of Botany, University of Newcastle upon Tyne” (Received 11 May 1965, accepted 16 September 1965) SUMMARY Anthranilic and o-nitrobenzoic acids act as mutual inhibitors of both growth and substrate oxidation for Nocardia opaca and a flavobacterium which can utilize either substance as sole source of carbon, nitrogen and energy, Growth of the former bacterium on anthranilate induced, appar- ently simultaneously, both the transport system for anthranilate uptake and the enzymic mechanism necessary for its complete oxidation to CO, and NH3. Among the enzymes induced by anthranilate was the complete sequence that oxidizes catechol to /3-oxoadipate; this was absent from organisms grown in fumarate or glucose media. The properties of the first enzyme in this sequence, a catechol-l,2-oxygenase, differ in several features from those of the same enzyme induced in this bacterium by growth on o-nitrobenzoic acid. INTRODUCTION Anthranilic (o-aminobenzoic) acid is an important intermediary metabolite in both biosynthetic and catabolic pathways in micro-organisms. It serves for in- stance as a precursor for tryptophan in Aerobacter aerogews and Escherichia coli (Doy & Gibson, 1961), in Nmrospora CT~SSQ(Yanofsky, 1956; Yanofsky & Rach- meler, 1958) and in saccharomyces mutants (Lingens, Hildinger & Hellman, 1958). Hydroxylation of anthranilic acid, in the 3-position, has been observed with rat liver preparations (Wiss & Hellman, 1953) but no hydroxylation of anthranilic acid has been conclusively demonstrated in micro-organisms. -

Chemicals Required for the Illicit Manufacture of Drugs Table 1 SUBSTANCES in TABLES I and II of the 1988 CONVENTION
Chemicals Required 1. A variety of chemicals are used in the illicit manufacture of for the Illicit drugs. The United Nations Convention against Illicit Traffic in Manufacture of Drugs Narcotic Drugs and Psychotropic Substances of 1988 (1988 Convention) refers to “substances frequently used in the illicit manufacture of narcotic drugs and psychotropic substances”. Twenty-two such substances are listed in Tables I and II of the 1988 Convention as in force on 1st May, 1998. (See Table1) Table 1 Table I Table II SUBSTANCES IN N-Acetylanthranilic acid. Acetic anhydride TABLES I AND II OF Ephedrine Acetone THE 1988 Ergometrine Anthranilic acid CONVENTION Ergotamine Ethyl ether Isosafrole Hydrochloric acid* Lysergic acid Methyl ethyl ketone 3,4-methylenedioxyphenyl-2-propanone Phenylacetic acid 1-phenyl-2-propanone Piperidine Piperonal Potassium permanganate Pseudoephedrine Sulphuric acid* Safrole Toluene The salts of the substances in this Table The salts of the substances whenever the existence of such salts is in this Table whenever the possible. existence of such salts is possible. * The salts of hydrochloric acid and sulphuric acid are specifically excluded from Table II. U N D C P 11 The term “precursor” is used to indicate any of these substances in the two Tables. Chemicals used in the illicit manufacture of narcotic drugs and psychotropic substances are often described as precursors or essential chemicals, and these include true precursors, solvents, oxidising agents and other Chemicals used in the illicit manufacture of narcotic substances. Although the term is not drugs and psychotropic substances are often technically correct, it has become common described as precursors or essential chemicals, and practice to refer to all such substances as these include true precursors, solvents, oxidising “precursors”.