Ocean Drilling Program Technical Note 5
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Simple Spectrophotometric Method for the Determination Of
J Clin Pathol: first published as 10.1136/jcp.14.2.202 on 1 March 1961. Downloaded from 202 Technical methods usual laboratory filters may give falsely increased values.) Of the clear and colourless filtrate 10 ml. is pipetted into A a 25 ml. measuring flask and 10 ml. reagent (3) added. simple spectrophotometric After mixing dilute with distilled water to the mark and method for the determination of renew mixing. After 10 minutes the mixture is ready for reading. The colour is stable, according to Watt and urea in blood and urine Chrisp, for 11 days. READING T. K. WITH, TOVE DREYER PETERSEN, AND BIRGIT PETERSEN From the Central Laboratory, Svend- The reactions are read in a spectrophotometer at 420 nm. borg County Hospital, Denmark with a blank prepared in the same way as the reaction mixture but with 3 ml. of distilled water instead of 3 ml. of blood. The urea standard (reagent 4) is treated in the Several methods for urea in determining blood and urine same way as the blood. Cuvettes of 1 cm. thickness are are in use in clinical laboratories, but none is ideal in suitable. mass routine The analysis. spectrophotometric method If p, s, and b are the extinctions of the the based on a modified Ehrlich sample, reagent proposed by Watt standard, and the blank respectively, the urea concen- and Chrisp (1954) for pure solutions is therefore of tration is interest because it can be modified for use in mass clinical analyses of blood and urine. Brown (1959) has l00(p-b)/(s-b) mg./100 ml. -

I. Direct Titration of Sulfate, II. High Precision Spectrophotometric Analysis Max Quentin Freeland Iowa State College
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1955 I. Direct titration of sulfate, II. High precision spectrophotometric analysis Max Quentin Freeland Iowa State College Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Analytical Chemistry Commons Recommended Citation Freeland, Max Quentin, "I. Direct titration of sulfate, II. High precision spectrophotometric analysis" (1955). Retrospective Theses and Dissertations. 14746. https://lib.dr.iastate.edu/rtd/14746 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMi films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -

SPECIAL SCIENTIFIC REPORT-FISHERIES Na 349
349 CHEMICAL ANALYSES OF MARINE AND ESTUARINE WATERS USED BY THE GALVESTON BIOLOGICAL LABORATORY SPECIAL SCIENTIFIC REPORT-FISHERIES Na 349 UNITED STATES DEPARTMENT OF THE INTERIOR FISH AND WILDLIFE SERVICE United States Department of the Interior, Fred A. Seaton, Secretary Fish and Wildlife Service, Arnie J. Suomela, Commissioner Bureau of Commercial Fisheries, Donald L, McKernan, Director CHEMICAL ANALYSES OF MARINE AND ESTUARINE WATERS USED BY THE GALVESTON BIOLOGICAL LABORATORY by Kenneth T. Marvin, Zoula P. Zein-Eldin, Billie Z. May and Larence M. Lansford Chemists Galveston, Texas United States Fish and Wildlife Service Special Scientific Report— Fisheries No. 349 Washington, D. C. June 1960 CONTENTS Introduction 1 Sample treatment prior to analysis 1 Sample storage containers 2 Analytical methods 2 Standard samples 2 Phosphate 3 Inorganic only 3 Total and inorganic 3 Total only 4 Nitrate-nitrite 5 Nitrite 5 Salinity 6 Copper 6 Sulfide 7 Oxygen 7 Total carbon dioxide 8 Ammonia 10 Chlorophyll 10 "Carbohydrates" 11 "Protein" (tyrosine equivalent) 12 Washing procedure for all analytical glassware 12 Literature cited 13 111 CHEMICAL AMLYSES OF MAP.INE AND ESTOARIKE mTERS USED BY TBE GALVESTON BIOLOGICAL lABORATORY by Kenneth T. Marvin, Zoula P. Zein-Eldin, Billie Z. May and Larence M. Lansford ABSTRACT This paper describes the chemical techniques and procedures used hy the Biological lahoratory of the U. S. Bureau of Commercial Jlsherles, Galveston^ Texas, for analyzing samples Involved In the chemical and hlo- logical survey of the marine and estuarlne waters of the Gulf of Mexico and also In the many laboratory and field studies and experiments that have heen made pertaining to the red tide investigation. -

Laboratory Equipment Used in Filtration
KNOW YOUR LAB EQUIPMENTS Test tube A test tube, also known as a sample tube, is a common piece of laboratory glassware consisting of a finger-like length of glass or clear plastic tubing, open at the top and closed at the bottom. Beakers Beakers are used as containers. They are available in a variety of sizes. Although they often possess volume markings, these are only rough estimates of the liquid volume. The markings are not necessarily accurate. Erlenmeyer flask Erlenmeyer flasks are often used as reaction vessels, particularly in titrations. As with beakers, the volume markings should not be considered accurate. Volumetric flask Volumetric flasks are used to measure and store solutions with a high degree of accuracy. These flasks generally possess a marking near the top that indicates the level at which the volume of the liquid is equal to the volume written on the outside of the flask. These devices are often used when solutions containing dissolved solids of known concentration are needed. Graduated cylinder Graduated cylinders are used to transfer liquids with a moderate degree of accuracy. Pipette Pipettes are used for transferring liquids with a fixed volume and quantity of liquid must be known to a high degree of accuracy. Graduated pipette These Pipettes are calibrated in the factory to release the desired quantity of liquid. Disposable pipette Disposable transfer. These Pipettes are made of plastic and are useful for transferring liquids dropwise. Burette Burettes are devices used typically in analytical, quantitative chemistry applications for measuring liquid solution. Differing from a pipette since the sample quantity delivered is changeable, graduated Burettes are used heavily in titration experiments. -

BLAUBRAND® Volumetric Instruments and Density Bottles
BLAUBRAND® Volumetric Instruments and Density Bottles Testing Instructions (SOP) March 2015 1. Introduction The standard DIN EN ISO 4787 describes both the design and the testing of the volumetric instruments of glass. The following Testing Instructions describe how to apply the ISO standard in practice. We recommend a testing every 1-3 years. The interval depends on the using of aggressive media and the cleaning procedure. These Instructions may also be used as a basis for the supervision of testing devices to DIN EN ISO 9001, DIN EN ISO 10012 and DIN EN ISO/IEC 17025. The test of the Density Bottles was effected on the basis of DIN EN ISO 4787. Meniscus adjustment with BLAUBRAND® Volumetric Instruments Meniscus adjustment Meniscus adjustment with ring mark with Schellbach stripe Read at the lowest point of the meniscus. Read at the point where the two arrows touch. Meniscus adjustment reflexion of liquid surface meniscus ring mark dark paper (p.e. black, blue) 2 2. Preparation for testing 2. Clear identification of the volumetric instrument to be tested Batch number, individual serial number, trademark, nominal volume and tolerances are directly printed ⇒ The test starts with a clear identification of the on every BLAUBRAND volumetric instruments. volumetric instrument in the test record. 2.1 Copy Test Record (see page 13) 2.2 Serial number/Identification number ⇒ Enter into Test Record All BLAUBRAND® volumetric instruments always carry a batch number, e.g., 13.04, or an individual serial number in the case of individual certificates, e.g., 13.040371 (year of production 2013, Batch No. -

Signature Redacted Department of Metallurgy Gau, R E Da Signature of Professor in Charge of Research
THERMODYNAMIC PROPERTIES OF THE GROUP VIa SULFIDES: CrS, Mo2 S3 AND WS2 by JOHN PATRICK HAGER B. S., Montana School of Mines (1958) M. S. , School of Mines and Metallurgy University of Missouri (1960) Submitted in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF SCIENCE at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY August, 1969 Signature of Author Signature redacted Department of Metallurgy gau, r e da Signature of Professor in Charge of Research Signature of Chairman of Departmental Committee Graduate Research nchives Signature redacted INST. 7 DEC 2 1969 4IBRARIES -~ U ii THERMODYNAMIC PROPERTIES OF THE GROUP VIa SULFIDES: CrS, Mo 2S 3 and WS 2 by John Patrick Hager Submitted to the Department of Metallurgy on August 18, 1969, in partial fulfillment of the requirements for the degree of Doctor of Science. ABSTRACT The thermodynamic properties of the metal-saturated phase of the Group VIa sulfides have been determined through a study of the effect of gas composition on the sulfidizing action of (H2S g) + H 2 (g) ) mixtures passed over heated metal samples. The following equations for the standard free energy of formation of CrS (c) ' Mo2S3(c) and WS2(c) were obtained: AFCc) ( 250) = -48,190 ( 760) + 13.27( 0.52)T; cal/l/2 g-mole S2(g) (1375-1570 0 K) AF 0 ( 220) = -41,730( 890) + 17.39( 0.59)1R; cal/l/2 g-mole S2(g) 102S3 (c)2(g (1365-1610 0 K) AF ( 220) = -40,110( 920) + 18.64( 0.63)T; cal/l/2 g-mole S WS 2 (c) 2(g) (1370-15650K) An additional study of the thermodynamic properties of Cu2S ) pro- vided a means of evaluating the experimental technique. -

Accuracy in Volume Measurements
Activity 1: Accuracy in volume measurements Requirements ● 50 mL burette ● Water ● Burette clamp and stand ● 100 mL graduated cylinder ● Funnel ● 100 mL beaker Health and safety, and outline instructions 1. Set up a burette on a stand assembly (see Figure 1). Figure 1 2. Fill the empty burette with water. Make sure the meniscus of the water in the burette rests on the 0.00 mL line at the top of the burette (see Figure 2). 48 48 48 48 35 35 35 35 49 49 49 49 36 36 36 36 50 50 50 50 0 0 0 0 37 37 37 37 1 1 1 1 38 38 38 38 2 2 2 2 39 39 39 39 3 3 3 3 40 40 40 40 0 0 4 0 4 0 4 4 41 41 41 41 1 1 1 1 2 2 5 2 5 2 5 5 42 42 42 42 3 3 3 3 4 4 6 4 6 4 6 6 43 43 43 43 5 5 5 5 6 6 7 6 7 6 7 7 44 44 44 44 7 7 7 7 8 8 8 8 8 8 8 8 45 45 45 45 9 9 9 9 10 10 9 10 9 10 9 9 46 46 46 46 Figure 2 Closed ClosedClosedClosedClosedClosedOpenClosedOpenClosedClosedOpenClosedOpen Closed Closed 3. Place a 100 mL graduated cylinder beneath the tip of the burette. Open the stopcock and slowly release about 40 mL of water from the burette into the graduated cylinder. Record the exact volume of water dispensed from the burette in the appropriate column of the results table. -

Mélanie Burette
Mélanie Burette French, born 16th June 1993 [email protected] [email protected] Position: Post-doctoral researcher (since January 2021) CRBM CNRS UMR5237 Montpellier, France Supervisor : Dr. Cécile Gauthier-Rouvière Academic achievements 2020: Doctor of Philosophy in Cell Biology Université de Montpellier, France IRIM CNRS UMR9004 Montpellier, France Supervisor : Dr. Matteo Bonazzi 2017: Master of Science in Microbiology and Immunology Université de Montpellier, France. 2015: Bachelor of Science in Ecology and Organismal Biology Université de Montpellier, France. 2011: A-level in Science Lycée Jean Renoir, France Awards and honors 2019: IRIM Institute Meeting Grant 2019: European Society of Endocrinoloy Meeting Grant for the 44th symposium on Hormones and Cell Regulation. 2018: Winner “Prix du meilleur poster” of the 16th meeting of PhD students in Chemical and Biological Sciences for Health CBS2 Doctoral School, Montpellier, France. List of Publications 1. Burette M. *, Bienvenu A.*, Inamdar K., Bordignon B., Swain J., Muriaux D., Martinez E., Bonazzi M. (Submitted) Identification of a new Coxiella lipid- binding effector protein by a vacuolar lipid profiling. Page 1 of 4 2. Martinez E., Huc-Brandt S., Brelle S., Allombert J., Cantet F., Gannoun-Zaki L., BURETTE M., Martin M., Letourneur F., Bonazzi M. and Molle V. (2020) The Coxiella burnetii secreted protein kinase CstK influences vacuole development and interacts with the GTPase-activating protein TBC1D5. Journal of Biological Chemistry, 295(21): 7391-7403, doi: 10.1074/jbc.RA119.01011. 3. Burette M. *, Allombert J. *, Lambou K., Maarifi G., Nisole S., Case ED., Blanchet FP., Hassen-Khodja C., Cabantous S., Samuel J., Martinez E., Bonazzi M. -

Titrations Titrations Are Done Often to Find out the Concentration of One Substance by Reacting It with Another Substance of Known Concentration
Titrations Titrations are done often to find out the concentration of one substance by reacting it with another substance of known concentration. They are often done with neutralisation reactions, but can be done with redox reactions. One substance (generally the one we don’t know the concentration) is put in the conical flask. It is measured using a volumetric burette pipette. The other substance is placed in the burette However, the standard phrase: titrate solution A with solution B means that A should be in the conical conical flask and B should be in the burette. flask A conical flask is used in preference to a beaker because it is easier to swirl the mixture in a conical flask without spilling the contents. pipette Method for Titration Make sure bottom of Using the pipette meniscus is on line on neck of pipette •rinse pipette with substance to go in it (often alkali). •pipette 25 cm3 of solution A into conical flask. The volumetric pipette will have a mark on its neck to show the level to fill to. The bottom of the meniscus should sit on this line. •touch surface of solution with pipette ( to ensure correct amount is added). A small amount of solution will be left in the pipette at this stage. The calibration of the pipette will take into account this effect. It should not be forced out. N Goalby chemrevise.org 1 Using the burette The burette should be rinsed out with substance that will be put in it. If it is not rinsed out the acid or alkali added may be diluted by residual water in the burette or may react with substances left from a previous titration. -

CHM 130 - Accuracy and the Measurement of Volume
CHM 130 - Accuracy and the Measurement of Volume PURPOSE: The purpose of this experiment is to practice using various types of volume measuring apparatus, focusing on their uses and accuracy. DISCUSSION: Volume measuring apparatus come in several different designs – graduated cylinders, volumetric flasks, pipets, burets, etc. Each design has a different application and a different accuracy. We are going to study these applications and the accuracy of the designs. In general, the less accurate the apparatus is, the easier and faster it is to use. So if great accuracy is not needed, why not be practical and use the fast and easy apparatus. In an experiment, the measurements made using a volume measuring apparatus should be at least as accurate as all the other measurements made in the experiment. For this reason, it is important to know the accuracy of different apparatus that are available. There are two kinds of errors in measurements. ACCURACY is the error associated with how close a measurement is to the true or actual value. If an instrument gives values that are very close to the true value we say that it is ACCURATE. Example: A graduated cylinder upon measuring the same sample three times gave 566 mL, 584 mL, and 541 mL. The average of these three values is 563.7 mL. If the true value was 563.688 mL, we would say that the average was accurate but the individual measurements were neither accurate nor precise. PRECISION is the error associated with how close several measurements of the same quantity are to each other. -
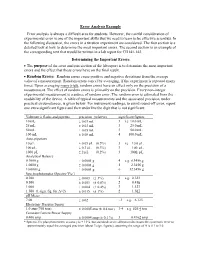
Error Analysis Example Error Analysis Is Always a Difficult Area for Students
Error Analysis Example Error analysis is always a difficult area for students. However, the careful consideration of experimental error is one of the important skills that we need to learn to be effective scientists. In the following discussion, the errors in a titration experiment are considered. The first section is a detailed look at how to determine the most important errors. The second section is an example of the corresponding text that would be written in a lab report for CH141-142. Determining the Important Errors The purpose of the error analysis section of the lab report is to determine the most important errors and the effect that those errors have on the final result. Random Errors: Random errors cause positive and negative deviations from the average value of a measurement. Random errors cancel by averaging, if the experiment is repeated many times. Upon averaging many trials, random errors have an effect only on the precision of a measurement. The effect of random errors is primarily on the precision. Every non-integer experimental measurement is a source of random error. The random error is estimated from the readability of the device. A table of typical measurements and the associated precision, under practical circumstances, is given below. For instrument readings, to avoid round-off error, report one extra significant figure and then underline the digit that is not significant. Volumetric flasks and pipettes precision (relative) significant figures 10 mL 0.03 mL 3 i.e. 10.0 mL 25 mL 0.03 mL 3 25.0 mL 50 mL 0.05 mL 3 50.0 mL 100 mL 0.08 mL 4 100.0 mL Auto-pipettors 10 L 0.05 L (0.5%) 3 i.e. -

Titrette Digital Bottle-Top Burette Leaflet
The next generation Digital Burette! Titrette® class A precision FIRST CLASS·BRAND With the Titrette® bottle-top burette, available in 10 ml, 25 ml, and 50 ml sizes, you can titrate quickly and reliably with highest precision, even in close quarters, with no power hookup needed – in the lab, in production, or in the field! I High accuracy Precise titration within the error limits of Class A I Smooth, low-force operation For sensitive, drop-wise titration I Compact design Light and stable I Easy handling No switching between 'fill' and 'titrate' I Simple maintenance Cleaning and part replacement right in the lab I PC interface (optional) Data transmission directly to the PC A closer look The control elements of the Titrette® bottle-top burette have an intuitive layout. Separate buttons for On/Off and Pause; CLEAR/Select button Clear button allows user to reset the display and select functions. Easy-grip hand wheel action and smooth precision gears for fast or drop-wise titration make handling extremely simple and reliable. PC interface (optional) (see ordering information) Digital display Replaceable batteries On/Off button Pause button Hand wheels Piston Inspection window Dispensing cylinder Titration and recirculating valve Valve block Freely rotating (Bottle thread GL 45) Titrating tube horizontally and vertically adjustable, with integrated discharge valve Recirculating tube Telescoping filling tube Screw cap Extras Four additional helpful electronic functions make your work easier. Hold down the CLEAR button to select the desired function: Adjustment with Calibration Easy Calibration schedule With Easy Calibration To save a date for the technology, you can next calibration, simply easily make calibra- store it under 'GLP'.