Mtor Links Incretin Signaling to HIF Induction in Pancreatic Beta Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Hypertension and Prolonged Vasoconstrictor Signaling in RGS2- Deficient Mice
Amendment history: Addendum (April 2003) Hypertension and prolonged vasoconstrictor signaling in RGS2- deficient mice Scott P. Heximer, … , Robert P. Mecham, Kendall J. Blumer J Clin Invest. 2003;111(4):445-452. https://doi.org/10.1172/JCI15598. Article Cardiology Signaling by hormones and neurotransmitters that activate G protein–coupled receptors (GPCRs) maintains blood pressure within the normal range despite large changes in cardiac output that can occur within seconds. This implies that blood pressure regulation requires precise kinetic control of GPCR signaling. To test this hypothesis, we analyzed mice deficient in RGS2, a GTPase-activating protein that greatly accelerates the deactivation rate of heterotrimeric G proteins in vitro. Both rgs2+/– and rgs2–/– mice exhibited a strong hypertensive phenotype, renovascular abnormalities, persistent constriction of the resistance vasculature, and prolonged response of the vasculature to vasoconstrictors in vivo. Analysis of P2Y receptor–mediated Ca2+ signaling in vascular smooth muscle cells in vitro indicated that loss of RGS2 increased agonist potency and efficacy and slowed the kinetics of signal termination. These results establish that abnormally prolonged signaling by G protein–coupled vasoconstrictor receptors can contribute to the onset of hypertension, and they suggest that genetic defects affecting the function or expression of RGS2 may be novel risk factors for development of hypertension in humans. Find the latest version: https://jci.me/15598/pdf Hypertension and prolonged See the related Commentary beginning on page 441. vasoconstrictor signaling in RGS2-deficient mice Scott P. Heximer,1 Russell H. Knutsen,1 Xiaoguang Sun,1 Kevin M. Kaltenbronn,1 Man-Hee Rhee,1 Ning Peng,2 Antonio Oliveira-dos-Santos,3 Josef M. -

Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: a Role for Histone Deacetylase 4
The Journal of Neuroscience, November 12, 2014 • 34(46):15327–15339 • 15327 Cellular/Molecular Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: A Role for Histone Deacetylase 4 Yelin Chen,1 Yuanyuan Wang,1 Zora Modrusan,3 Morgan Sheng,1 and Joshua S. Kaminker1,2 Departments of 1Neuroscience, 2Bioinformatics and Computational Biology, and 3Molecular Biology, Genentech Inc., South San Francisco, California 94080 Neuronal gene expression is modulated by activity via calcium-permeable receptors such as NMDA receptors (NMDARs). While gene expression changes downstream of evoked NMDAR activity have been well studied, much less is known about gene expression changes that occur under conditions of basal neuronal activity. In mouse dissociated hippocampal neuronal cultures, we found that a broad NMDAR antagonist, AP5, induced robust gene expression changes under basal activity, but subtype-specific antagonists did not. While some of the gene expression changes are also known to be downstream of stimulated NMDAR activity, others appear specific to basal NMDARactivity.ThegenesalteredbyAP5treatmentofbasalcultureswereenrichedforpathwaysrelatedtoclassIIahistonedeacetylases (HDACs), apoptosis, and synapse-related signaling. Specifically, AP5 altered the expression of all three class IIa HDACs that are highly expressed in the brain, HDAC4, HDAC5, and HDAC9, and also induced nuclear accumulation of HDAC4. HDAC4 knockdown abolished a subset of the gene expression changes induced by AP5, and led to neuronal death under -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Figure S1. HAEC ROS Production and ML090 NOX5-Inhibition
Figure S1. HAEC ROS production and ML090 NOX5-inhibition. (a) Extracellular H2O2 production in HAEC treated with ML090 at different concentrations and 24 h after being infected with GFP and NOX5-β adenoviruses (MOI 100). **p< 0.01, and ****p< 0.0001 vs control NOX5-β-infected cells (ML090, 0 nM). Results expressed as mean ± SEM. Fold increase vs GFP-infected cells with 0 nM of ML090. n= 6. (b) NOX5-β overexpression and DHE oxidation in HAEC. Representative images from three experiments are shown. Intracellular superoxide anion production of HAEC 24 h after infection with GFP and NOX5-β adenoviruses at different MOIs treated or not with ML090 (10 nM). MOI: Multiplicity of infection. Figure S2. Ontology analysis of HAEC infected with NOX5-β. Ontology analysis shows that the response to unfolded protein is the most relevant. Figure S3. UPR mRNA expression in heart of infarcted transgenic mice. n= 12-13. Results expressed as mean ± SEM. Table S1: Altered gene expression due to NOX5-β expression at 12 h (bold, highlighted in yellow). N12hvsG12h N18hvsG18h N24hvsG24h GeneName GeneDescription TranscriptID logFC p-value logFC p-value logFC p-value family with sequence similarity NM_052966 1.45 1.20E-17 2.44 3.27E-19 2.96 6.24E-21 FAM129A 129. member A DnaJ (Hsp40) homolog. NM_001130182 2.19 9.83E-20 2.94 2.90E-19 3.01 1.68E-19 DNAJA4 subfamily A. member 4 phorbol-12-myristate-13-acetate- NM_021127 0.93 1.84E-12 2.41 1.32E-17 2.69 1.43E-18 PMAIP1 induced protein 1 E2F7 E2F transcription factor 7 NM_203394 0.71 8.35E-11 2.20 2.21E-17 2.48 1.84E-18 DnaJ (Hsp40) homolog. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

Supplementary Table 1
Supplementary Table 1. 492 genes are unique to 0 h post-heat timepoint. The name, p-value, fold change, location and family of each gene are indicated. Genes were filtered for an absolute value log2 ration 1.5 and a significance value of p ≤ 0.05. Symbol p-value Log Gene Name Location Family Ratio ABCA13 1.87E-02 3.292 ATP-binding cassette, sub-family unknown transporter A (ABC1), member 13 ABCB1 1.93E-02 −1.819 ATP-binding cassette, sub-family Plasma transporter B (MDR/TAP), member 1 Membrane ABCC3 2.83E-02 2.016 ATP-binding cassette, sub-family Plasma transporter C (CFTR/MRP), member 3 Membrane ABHD6 7.79E-03 −2.717 abhydrolase domain containing 6 Cytoplasm enzyme ACAT1 4.10E-02 3.009 acetyl-CoA acetyltransferase 1 Cytoplasm enzyme ACBD4 2.66E-03 1.722 acyl-CoA binding domain unknown other containing 4 ACSL5 1.86E-02 −2.876 acyl-CoA synthetase long-chain Cytoplasm enzyme family member 5 ADAM23 3.33E-02 −3.008 ADAM metallopeptidase domain Plasma peptidase 23 Membrane ADAM29 5.58E-03 3.463 ADAM metallopeptidase domain Plasma peptidase 29 Membrane ADAMTS17 2.67E-04 3.051 ADAM metallopeptidase with Extracellular other thrombospondin type 1 motif, 17 Space ADCYAP1R1 1.20E-02 1.848 adenylate cyclase activating Plasma G-protein polypeptide 1 (pituitary) receptor Membrane coupled type I receptor ADH6 (includes 4.02E-02 −1.845 alcohol dehydrogenase 6 (class Cytoplasm enzyme EG:130) V) AHSA2 1.54E-04 −1.6 AHA1, activator of heat shock unknown other 90kDa protein ATPase homolog 2 (yeast) AK5 3.32E-02 1.658 adenylate kinase 5 Cytoplasm kinase AK7 -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Human Ortholog of Drosophila Melted Impedes SMAD2 Release from TGF
Human ortholog of Drosophila Melted impedes SMAD2 PNAS PLUS release from TGF-β receptor I to inhibit TGF-β signaling Premalatha Shathasivama,b,c, Alexandra Kollaraa,c, Maurice J. Ringuetted, Carl Virtanene, Jeffrey L. Wranaa,f, and Theodore J. Browna,b,c,1 aLunenfeld-Tanenbaum Research Institute at Mount Sinai Hospital, Toronto, ON, Canada M5T 3H7; Departments of bPhysiology, cObstetrics and Gynaecology, dCell and Systems Biology, and fMolecular Genetics, University of Toronto, Toronto, ON, Canada M5S 3G5; and ePrincess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada M5G 1L7 Edited by Igor B. Dawid, The Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, and approved May 5, 2015 (received for review March 11, 2015) Drosophila melted encodes a pleckstrin homology (PH) domain- The gene locus encompassing human VEPH1, 3q24-25, lies containing protein that enables normal tissue growth, metabo- within a region frequently amplified in ovarian cancer (6, 7). Tan lism, and photoreceptor differentiation by modulating Forkhead et al. (8) found that this locus was also amplified in 7 of 12 ep- box O (FOXO), target of rapamycin, and Hippo signaling pathways. ithelial ovarian cancer cell lines. A gene copy number analysis of Ventricular zone expressed PH domain-containing 1 (VEPH1) is the 68 primary tumors by Ramakrishna et al. (9) identified frequent mammalian ortholog of melted, and although it exhibits tissue- (>40%) VEPH1 gene amplification that correlated with tran- restricted expression during mouse development and is poten- script levels. We determined the impact of VEPH1 on gene ex- tially amplified in several human cancers, little is known of its pression in an ovarian cancer cell line using a whole-genome function. -
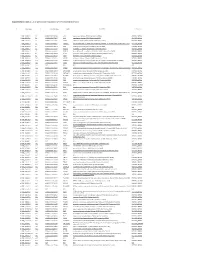
Supplemetary Table 2. List of Genes Down-Regulated in LPAR6 Knocked Down Cells
Supplemetary Table 2. List of genes down-regulated in LPAR6 knocked down cells g# initial alias c# converted alias name description namespace 1 NM_002317.5 1.1 ENSG00000113083 LOX lysyl oxidase [Source:HGNC Symbol;Acc:6664] REFSEQ_MRNA 2 NM_006183.4 2.1 ENSG00000133636 NTS neurotensin [Source:HGNC Symbol;Acc:8038] REFSEQ_MRNA 3 NM_005213.3 3.1 ENSG00000121552 CSTA cystatin A (stefin A) [Source:HGNC Symbol;Acc:2481] REFSEQ_MRNA 4 NM_007231.3 4.1 ENSG00000087916 SLC6A14 solute carrier family 6 (amino acid transporter), member 14 [Source:HGNC Symbol;Acc:11047] REFSEQ_MRNA 5 NM_001873.2 5.1 ENSG00000109472 CPE carboxypeptidase E [Source:HGNC Symbol;Acc:2303] REFSEQ_MRNA 6 NM_019856.1 6.1 ENSG00000101605 MYOM1 myomesin 1, 185kDa [Source:HGNC Symbol;Acc:7613] REFSEQ_MRNA 7 NM_032590.4 7.1 ENSG00000089094 KDM2B lysine (K)-specific demethylase 2B [Source:HGNC Symbol;Acc:13610] REFSEQ_MRNA 8 NM_001901.2 8.1 ENSG00000118523 CTGF connective tissue growth factor [Source:HGNC Symbol;Acc:2500] REFSEQ_MRNA 9 NM_007183.2 9.1 ENSG00000184363 PKP3 plakophilin 3 [Source:HGNC Symbol;Acc:9025] REFSEQ_MRNA 10 NM_182965.2 10.1 ENSG00000176170 SPHK1 sphingosine kinase 1 [Source:HGNC Symbol;Acc:11240] REFSEQ_MRNA 11 NM_152423.4 11.1 ENSG00000157502 MUM1L1 melanoma associated antigen (mutated) 1-like 1 [Source:HGNC Symbol;Acc:26583] REFSEQ_MRNA 12 NM_002923.3 12.1 ENSG00000116741 RGS2 regulator of G-protein signaling 2, 24kDa [Source:HGNC Symbol;Acc:9998] REFSEQ_MRNA 13 NR_003038.2 13.1 N/A N/A N/A N/A 14 NM_080862.1 14.1 ENSG00000175093 SPSB4 splA/ryanodine receptor -

CHIP Phosphorylation by Protein Kinase G Enhances Protein Quality Control and Attenuates Cardiac Ischemic Injury
ARTICLE https://doi.org/10.1038/s41467-020-18980-x OPEN CHIP phosphorylation by protein kinase G enhances protein quality control and attenuates cardiac ischemic injury Mark J. Ranek1, Christian Oeing1, Rebekah Sanchez-Hodge3, Kristen M. Kokkonen-Simon 1, Danielle Dillard1, M. Imran Aslam1, Peter P. Rainer 1,6, Sumita Mishra1, Brittany Dunkerly-Eyring1, Ronald J. Holewinski2, Cornelia Virus3, Huaqun Zhang5, Matthew M. Mannion5, Vineet Agrawal1, Virginia Hahn1, Dong I. Lee1, Masayuki Sasaki1, Jennifer E. Van Eyk 2, Monte S. Willis3, Richard C. Page 5, Jonathan C. Schisler 3,4 & ✉ David A. Kass 1 1234567890():,; Proteotoxicity from insufficient clearance of misfolded/damaged proteins underlies many diseases. Carboxyl terminus of Hsc70-interacting protein (CHIP) is an important regulator of proteostasis in many cells, having E3-ligase and chaperone functions and often directing damaged proteins towards proteasome recycling. While enhancing CHIP functionality has broad therapeutic potential, prior efforts have all relied on genetic upregulation. Here we report that CHIP-mediated protein turnover is markedly post-translationally enhanced by direct protein kinase G (PKG) phosphorylation at S20 (mouse, S19 human). This increases CHIP binding affinity to Hsc70, CHIP protein half-life, and consequent clearance of stress- induced ubiquitinated-insoluble proteins. PKG-mediated CHIP-pS20 or expressing CHIP- S20E (phosphomimetic) reduces ischemic proteo- and cytotoxicity, whereas a phospho- silenced CHIP-S20A amplifies both. In vivo, depressing PKG activity lowers CHIP-S20 phosphorylation and protein, exacerbating proteotoxicity and heart dysfunction after ischemic injury. CHIP-S20E knock-in mice better clear ubiquitinated proteins and are cardio- protected. PKG activation provides post-translational enhancement of protein quality control via CHIP.