UVA Chemical Filters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Booklet
www.mmsus.com | 952.525.2005 COVID-19 PRODUCTS Midwest Mechanical Solutions is your partner in optimizing your building for a safe, healthy, and comfortable environment for all. Contact your MMS Representative at any time, we are here to help. Advanced HEPA Filtration Recirculation/Negative Pressure Units AF400NP 1400 cubic foot room / 280 CFM • Air Flow: Nominal 132 l/s / 280 CFM • Voltage: 120V (2A) or 230V (1A) • Dimensions: 21"H x 13"W x 16"D / 55cm x 33cm x 40cm • Filtration: 2” Pre-lter MERV10 6” Medical grade HEPA lter 99.97% (to 0.3 micron) • Optional: chemical lter • Inlet: Bottom inlet • Discharge: 4” Discharge collar • Exhaust kit included • Sound Level: Whisper quiet, 56 dBA • Controls: Remote ON/OFF, communication capabilities, audible and visual alarms AF1000NP 3000 cubic foot room / 600 CFM • Air Flow: Nominal 283 l/s / 600 CFM • Voltage: 120V (2A) or 230V (1A) • Dimensions: 56"H x 16"W x 22"D / 132cm x 40cm x 56cm • Filtration: 2” Pre-lter MERV10 12” High capacity pleated lter MERV12 6” Medical grade HEPA lter 99.97% (down to 0.3 micron) • Optional: chemical lter (up to 50 lbs./23 kg) • Inlet: Bottom inlet • Discharge: 6” Discharge collar (4”, 8” optional) • Exhaust kit included • Sound Level: Whisper quiet, 54 dBA Controls: • Remote ON/OFF, communication capabilities, audible and visual alarms AF2000NP 5000 cubic foot room / 1000 CFM • Air Flow: Nominal 283 l/s / 600 CFM • Voltage: 120V (2A) or 230V (1A) • Dimensions: 56"H x 16"W x 22"D / 132cm x 40cm x 56cm • Filtration: 2” Pre-lter MERV10 12” High capacity pleated lter -

A Thesis Entitled Evaluating UVB and UVA Boosting Technologies For
A Thesis entitled Evaluating UVB and UVA Boosting Technologies for Chemical and Physical Sunscreens by An Ngoc Hiep Huynh Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Master of Science Degree in Pharmaceutical Sciences Industrial Pharmacy ___________________________________________ Gabriella Baki, Ph.D., Committee Chair ___________________________________________ Jerry Nesamony, Ph.D., Committee Member ___________________________________________ Matthew W. Liberatore, Ph.D., Committee Member ___________________________________________ Dr. Amanda C. Bryant-Friedrich, Dean College of Graduate Studies The University of Toledo May 2020 Copyright 2020 An Ngoc Hiep Huynh This document is copyrighted material. Under copyright law, no parts of this document may be reproduced without the expressed permission of the author. An Abstract of Evaluating UVB and UVA Boosting Technologies for Chemical and Physical Sunscreens by An Ngoc Hiep Huynh Submitted to the Graduate Faculty as partial fulfillment of the requirements for the Master of Science Degree in Pharmaceutical Sciences Industrial Pharmacy The University of Toledo May 2020 There are currently 14 organic and 2 inorganic UV filters approved in the United States. Due to coral reef safety concerns, octinoxate and oxybenzone have been banned in Hawaii, Key West, FL and the US Virgin Islands; and octocrylene is also being studied for its potential impact on coral reef safety, leaving 11 organic UV filters as viable options for sunscreen manufacturers – with limitations on their combination. Since consumers are always looking for sunscreens with high SPF and broad-spectrum protection, the need for UVB and UVA protection boosting technologies is greater than ever. In a preliminary study, about two dozen emollients were scanned for their SPF boosting capability with selected organic UV filters. -

GAO-18-61, SUNSCREEN: FDA Reviewed Applications For
United States Government Accountability Office Report to Congressional Committees November 2017 SUNSCREEN FDA Reviewed Applications for Additional Active Ingredients and Determined More Data Needed GAO-18-61 November 2017 SUNSCREEN FDA Reviewed Applications for Additional Active Ingredients and Determined More Data Needed Highlights of GAO-18-61, a report to congressional committees Why GAO Did This Study What GAO Found Using sunscreen as directed with other The Food and Drug Administration (FDA), within the Department of Health and sun protective measures may help Human Services, implemented requirements for reviewing applications for reduce the risk of skin cancer—the sunscreen active ingredients within time frames set by the Sunscreen Innovation most common form of cancer in the Act, which was enacted in November 2014. For example, the agency issued a United States. In the United States, guidance document on safety and effectiveness testing in November 2016. sunscreen is considered an over-the- counter drug, which is a drug available As of August 2017, all applications for sunscreen active ingredients remain to consumers without a prescription. pending after the agency determined more safety and effectiveness data are Some sunscreen active ingredients not needed. By February 2015, FDA completed its initial review of the safety and currently marketed in the United States effectiveness data for each of the eight pending applications, as required by the have been available in products in act. FDA concluded that additional data are needed to determine that the other countries for more than a ingredients are generally recognized as safe and effective (GRASE), which is decade. Companies that manufacture needed so that products using the ingredients can subsequently be marketed in some of these ingredients have sought the United States without FDA’s premarket approval. -

Toxic Effects
Chapter 8 Toxic effects Toxic and other adverse effects studies of tens or hundreds of patients spectrum to which the skin is exposed of sunscreens (Thune, 1984; English et al., 1987; (Gasparro et al., 1998). Since UVB is the In order for a sunscreen to have a toxic Lenique et al., 1992; Szczurko et al., primary stimulus for adaptation of the effect on living tissues, it must penetrate 1994; Trevisi et al., 1994; Gonçalo et al., skin to sunlight, less adaptation might be the skin. There is some evidence that 1995; Ang et al., 1998) and reviews expected to develop in individuals who this can occur (see p. 63 et seq.). (Dromgoole & Maibach, 1990; Gonzalez use sunscreens regularly. The adaptive & Gonzalez, 1996; Schauder & Ippen, responses include thickening of the epi- Human studies 1997). In the past, PABA and its esters dermis and transfer of melanin-contain- No published studies of toxic effects in were the most commonly reported ing granules to keratinocytes (tanning) humans were available to the Working contact and photoallergens in sun- (Fig. 44), which reduces the trans- Group. screens (Funk et ai., 1997), and this find- parency of the skin to UVA and UVB ing contributed to a reduction in their use (Fusaro et al., 1966; Olson et al., 1973). Contact sensitivity in sunscreens. The contact or photocon- Several reports showed that UVR- There are numerous reports of cases of tact allergen in sunscreens most induced injury, such as dermal connec- allergic reactions and photoreactivity to frequently cited today is benzophenone- tive tissue damage and sunburn cell for- sunscreens, but the prevalence of this 3, followed by dibenzoyl methanes. -
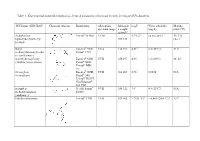
Table 1 - Experimental and Predicted Physical-Chemical Parameters of the Most Recently Investigated UV-Absorbers
Table 1 - Experimental and predicted physical-chemical parameters of the most recently investigated UV-absorbers. INCI name (INN/XAN) Chemical structure Brand name Absorption Molecula LogP Water solubility Melting spectrum range r weight (mg/L) point (°C) (g/mol)4 diethylamino Uvinul® A Plus UVA1 5.7-6.21 <0.01 (20°C) 1 54; 314 hydroxybenzoyl hexyl 397.515 (dec.) 1 benzoate Butyl Eusolex® 9020, UVA 310.393 4.514 2.2 (25°C)4 83.54 methoxydibenzoylmetha Parsol® 1789 ne (avobenzone) 4-methylbenzylidene Eusolex® 6300 UVB 258.397 4.95 1.3 (20°C) 66–68 camphor (enzacamene) Parsol® 5000 Uvinul® MBC 95 Octocrylene Eusolex® OCR, UVB 361.485 6.783 0.00383 N/A (octocrilene) Parsol® 340, Uvinul® N539T, NeoHeliopan® 303 USP isoamyl p- Neo Heliopan® UVB 248.322 3.61 4.9 (25°C)1 N/A methoxycinnamate E1000 (amiloxate) Ethylhexyl triazone Uvinul® T150 UVB 823.092 > 7(20 °C) 6 < 0.001 (20.0 °C) 6 1296 Ethylhexyl Parsol® MCX, UVB 290.403 6.14 0.041 (24 °C and N/A methoxycinnamate Heliopan® New pH 7.1) 4 (octinoxate) Ethylhexyl dimethyl Escalol™ 507 UVB 277.4084 5.774 0.54 (25 °C) 4 N/A PABA (padimate-O) Arlatone 507 Eusolex 6007 benzophenone-3 Eusolex® 4360 UVA2+ UVB 228.247 3.72 3.7 (20°C) 2 62-652 (oxybenzone) bis-ethylhexyloxyphenol Tinosorb® S UVA1+UVB 627.826 12.61 <10-4 80.401 methoxyphenol triazine (bemotrizinol) Phenylbenzimidazole Eusolex® 232 UVA2+ UVB 274.2945 -1.1 (pH 5) > 30% (As sodium N/A sulfonic acid Parsol® HS -2.1 (pH 8)5 or (ensulizole) Neo Heliopan® triethanolammoniu Hydro m salt at 20°C) 5 1 (3) 2 (34) 3 (44) 4 Pubchem 5 SCCP/1056/06 Opinion on phenylbenzimidazole sulfonic acid and its salts 6 BASF safety data sheet Table 2 – In vitro studies for the assessment of skin permeation/penetration of sunscreens. -

Sunscreen Enhancement of Octyl Methoxycinnamate Microcapsules by Using Two Biopolymers As Wall Materials
polymers Article Sunscreen Enhancement of Octyl Methoxycinnamate Microcapsules by Using Two Biopolymers as Wall Materials Chuntao Xu 1,2 , Xuemin Zeng 3, Zujin Yang 4,* and Hongbing Ji 1,3,4,5,* 1 School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China; [email protected] 2 School of Information Engineering, Zhongshan Polytechnic, Zhongshan 528400, China 3 Fine Chemical Industry Research Institute, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China; [email protected] 4 School of Chemical Engineering and Technology, Sun Yat-Sen University, Zhuhai 519082, China 5 School of Chemical Engineering, Guangdong University of Petrochemical Technology, Maoming 525000, China * Correspondence: [email protected] (Z.Y.); [email protected] (H.J.) Abstract: Octyl methoxycinnamate (OMC) is widely used as a chemical sunscreen in sunscreen cosmetics. However, its direct contact with the skin would bring certain risks, such as skin photo- sensitive reaction. How to improve the effect of skin photodamage protection has become a current research hotspot. Encapsulating ultraviolet (UV) filters into microcapsules is an interesting method to increase the photostability of filters. In this study, sodium caseinate (SC) and arabic gum (GA) are chosen as wall materials to prepare synergistic sunscreen microcapsules by complex coacervation technology. A series of experiments are conducted to investigate the effects of pH, wall material concentration, and wall/core ratio on the formation of OMC microcapsules. The morphology, com- position, and stability of OMC microcapsules are characterized by scanning electron microscopy Citation: Xu, C.; Zeng, X.; Yang, Z.; Ji, (SEM), Fourier transform infrared spectroscopy (FTIR), and thermogravimetric analysis (TGA). -

Photophysics and Skin Penetration of Active Agents in a Commercial Sunscreen and Insect Repellent
PHOTOPHYSICS AND SKIN PENETRATION OF ACTIVE AGENTS IN A COMMERCIAL SUNSCREEN AND INSECT REPELLENT by DONALD PRETTYPAUL A Dissertation submitted to the Graduate School-Newark Rutgers, The State University of New Jersey In partial fulfillment of the requirements for the degree of Doctor of Philosophy Graduate Program in Chemistry written under the direction of Professor Richard Mendelsohn Professor Piotr Piotrowiak and approved by Newark, New Jersey October 2018 ©2018 Donald Prettypaul ALL RIGHTS RESERVED ABSTRACT OF DISSERTATION PHOTOPHYSICS AND SKIN PENETRATION OF ACTIVE AGENTS IN A COMMERCIAL SUNSCREEN AND INSECT REPELLENT By DONALD PRETTYPAUL Dissertation co-Directors: Professor Richard Mendelsohn Professor Piotr Piotrowiak This dissertation is focused on active agents in commercial sunscreen and insect repellent products. It consists of two parts, the first focusing on the photophysics of a sunscreen active agent and the second on the permeation and spatial distribution of the sunscreen active and an insect repellent active when these agents are applied to ex-vivo human skin. In the photochemistry study, ultrafast spectroscopy was used to study the excited state dynamics of the sunscreen molecule, Bemotrizinol. The work focused on the dissipation rates of the electronic excitation energy in different solvents. To complement the results from time-resolved femtosecond spectroscopy, Hartree- Fock UH/UHF 6-31G* calculations were used to characterize the ground and excited states potential energy surfaces. The results indicate that the excited state deactivation pathway follows a proton coupled electron transfer process which ii proceeds via a concerted mechanism. The dependencies on solvent polarity, viscosity, and H/D isotope effects, were investigated. Sunscreen products have been developed to protect skin from ultraviolet (UV) radiation; to achieve adequate protection, the sunscreen must be evenly applied and remain on the surface of the skin. -

New Technology Provides Cosmetically Elegant Photoprotection Zoe Diana Draelos, MD; Christian Oresajo; Margarita Yatskayer; Angelike Galdi; Isabelle Hansenne
COSMETIC CONSULTATION New Technology Provides Cosmetically Elegant Photoprotection Zoe Diana Draelos, MD; Christian Oresajo; Margarita Yatskayer; Angelike Galdi; Isabelle Hansenne he primary preventable cause of photoaging is United States, as there is no official rating system for exposure to UVA radiation. This wavelength UVA photoprotection yet. T emitted by the sun is present year round in all Ecamsule absorbs UVA radiation in the range of 320 to latitudes. Currently, the majority of sun-protective prod- 360 nm, but its peak absorbance occurs at 345 nm. It is ucts provide excellent UVB protection with minimal UVA typically combined with other organic sunscreen ingredi- protection. Although UVB protection is important in ents, such as avobenzone and octocrylene. Avobenzone, order to prevent sun damage to the skin from occurring, which is a photounstable photoprotectant, becomes UVA protection is equally important. New developments photostable when combined with octocrylene. These in raw material science have led to the manufacture of ingredients yield a photostable broad-spectrum sunscreen novel ingredients able to provide unprecedented photo- combination. However, the active sunscreen agents are protection COSin the UVA spectrum. DERMonly part of the formulation. Also important in sunscreen One of the most significant developments in cutaneous is the construction of the vehicle to deliver the photo- UVA protection was the discovery of ecamsule (Figure 1), protectants in an aesthetically pleasing manner, enticing also known as Mexoryl SX. Mexoryl SX, which has patients to wear the product. Sunscreens fail to be effec- the International Nomenclature of Cosmetic Ingredients tive if they remain in the bottle. name of terephthalylidene dicamphor sulfonic acid, is In order to evaluate the efficacy and tolerability of a water soluble. -

1. Background and Action Requested
June 27, 2019 Dockets Management Staff (HFA-305) Food and Drug Administration 5630 Fishers Lane, Rm. 1061 Rockville, MD 20852 Re: Comments Submitted to Docket No. 1978N-0018 (Formerly Docket No. FDA–1978–N– 0038) for “Sunscreen Drug Products for Over-the-Counter Human Use,” Regulatory Information No. 0910-AF43 DSM Nutritional Products LLC (“DSM”) is pleased to provide the following comments in response to the Food and Drug Administration’s (FDA’s) Tentative Final Monograph (“TFM”) for Sunscreen Drug Products Over-the-Counter (OTC) Human Use, published at 84 Fed. Reg. 6204 (February 26, 2019). As a global science-based company active in Nutrition, Health and Sustainable Living, DSM is a world leading supplier of vitamins, feed, food, pharmaceutical, cosmetic, personal care and aroma ingredients. In sunscreens, DSM is a leading world-wide provider of numerous UV filters, including Avobenzone. DSM takes human health and safety very seriously and believes that sunscreens are proven and effective interventions that help provide consumer protection against skin cancers and skin damage caused by the sun’s rays. DSM is committed to proactively supporting the safety and availability of existing and new sunscreen active ingredients that help protect public health. DSM requests that FDA modify its proposed rule taking into consideration the comments provided herein. 1. Background and Action Requested Skin cancer is a significant, and largely preventable, public health concern. Sunscreens have been widely and safely used by the general public for their photoprotective properties, including prevention of photocarcinogenesis and photoaging and management of photodermatoses for more than 40 years. FDA’s Sunscreen Monograph rule has identified the permitted active ingredients (UV-filters) for sunscreens since 1978. -

FDA Proposes Sunscreen Regulation Changes February 2019
FDA Proposes Sunscreen Regulation Changes February 2019 The U.S. Food and Drug Administration (FDA) regulates sunscreens to ensure they meet safety and eectiveness standards. To improve the quality, safety, and eectiveness of sunscreens, FDA issued a proposed rule that describes updated proposed requirements for sunscreens. Given the recognized public health benets of sunscreen use, Americans should continue to use broad spectrum sunscreen with SPF 15 or higher with other sun protective measures as this important rulemaking eort moves forward. Highlights of FDA’s Proposals Sunscreen active ingredient safety and eectiveness Two ingredients (zinc oxide and titanium dioxide) are proposed to be safe and eective for sunscreen use and two (aminobenzoic acid (PABA) and trolamine salicylate) are 1 proposed as not safe and eective for sunscreen use. FDA proposes that it needs more safety information for the remaining 12 sunscreen ingredients (cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, padimate O, sulisobenzone, oxybenzone, avobenzone). New proposed sun protection factor Sunscreen dosage forms (SPF) and broad spectrum Sunscreen sprays, oils, lotions, creams, gels, butters, pastes, ointments, and sticks are requirements 2 proposed as safe and eective. FDA 3 • Raise the maximum proposed labeled SPF proposes that it needs more data for from SPF 50+ to SPF 60+ sunscreen powders. • Require any sunscreen SPF 15 or higher to be broad spectrum • Require for all broad spectrum products SPF 15 and above, as SPF increases, broad spectrum protection increases New proposed label requirements • Include alphabetical listing of active ingredients on the front panel • Require sunscreens with SPF below 15 to include “See Skin Cancer/Skin Aging alert” on the front panel 4 • Require font and placement changes to ensure SPF, broad spectrum, and water resistance statements stand out Sunscreen-insect repellent combination 5 products proposed not safe and eective www.fda.gov. -

WO 2013/036901 A2 14 March 2013 (14.03.2013) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2013/036901 A2 14 March 2013 (14.03.2013) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 8/30 (2006.01) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (21) International Application Number: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, PCT/US2012/054376 DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (22) International Filing Date: HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, 10 September 2012 (10.09.2012) KR, KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, (25) Filing Language: English NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, (26) Publication Language: English RW, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, (30) Priority Data: ZM, ZW. 61/532,701 9 September 201 1 (09.09.201 1) US (84) Designated States (unless otherwise indicated, for every (71) Applicant (for all designated States except US): UNIVER¬ kind of regional protection available): ARIPO (BW, GH, SITY OF FLORIDA RESEARCH FOUNDATION, GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, INC. -

OSEQUE ZSOLE DUAL SUN BLOCK- Octinoxate Avobenzone Bisoctrizole Spray SONGHAK CO., LTD
OSEQUE ZSOLE DUAL SUN BLOCK- octinoxate avobenzone bisoctrizole spray SONGHAK CO., LTD. ---------- Active ingredients: Octyl Methoxycinnamate 7.5%, Ethylhexyl Salicylate 5%, Avobenzone 2%, Bisoctrizole 0.5% Purpose: Sunscreen Inactive Ingredients: Water, Alcohol Denat, Butylene Glycol, Glycerin, Bis-Ethylhexyloxyphenol Methoxyphenyl Triazine, Niacinamide, Isononyl Isononanoate, Glacier Water, Butyloctyl Salicylate, Xanthan Gum, Decyl Glucoside, Disodium EDTA, Methylparaben, Propylparaben, Fragrance Do not use: on wounds, rashes, dermatitis or damaged skin Keep out of reach of children. In case of accidental ingestion, seek professional assistance or contact a Poison Control Center immediately Stop use: Please stop using this product and contact a dermatologist 1. If allergic reaction or irritation occurs 2. If direct sunlight affects the area as above When using this product: Do not use other than directed Warnings: For external use only. Not to be swallowed. Avoid contact with eyes. Discontinue use if signs of irritation or rash appear Uses: Spray or apply liberally over the face and body 15 minutes prior to sun exposure. Always ensure total coverage of all sun exposed areas. Re-apply every 1-2 hours and always after swimming. For spray type, shake it well before use and spray it evenly at 20cm-30cm apart from face or wherever needed. For spray type - shake it well before use For lotion type - turn a cap to use Storage: Keep in a dry or room temperature area OSEQUE ZSOLE DUAL SUN BLOCK octinoxate avobenzone bisoctrizole spray Product