Augment® Bone Graft
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Subtalar Joint Version 1.0 Effective June 15, 2021
CLINICAL GUIDELINES CMM-407: Arthroscopy: Subtalar Joint Version 1.0 Effective June 15, 2021 Clinical guidelines for medical necessity review of Comprehensive Musculoskeletal Management Services. © 2021 eviCore healthcare. All rights reserved. Comprehensive Musculoskeletal Management Guidelines V1.0 CMM-407: Arthroscopy: Subtalar Joint Definitions 3 General Guidelines 3 Indications 4 Non-Indications 5 Procedure (CPT®) Codes 5 References 6 ______________________________________________________________________________________________________ ©2021 eviCore healthcare. All Rights Reserved. Page 2 of 6 400 Buckwalter Place Boulevard, Bluffton, SC 29910 (800) 918-8924 www.eviCore.com Comprehensive Musculoskeletal Management Guidelines V1.0 Definitions Red flags indicate comorbidities that require urgent/emergent diagnostic imaging and/or referral for definitive therapy. Clinically meaningful improvement is defined as at least 50% improvement noted on global assessment. General Guidelines Either of the following are considered red flag conditions for subtalar joint arthroscopy: Post-reduction evaluation and management of the subtalar dislocation Septic arthritis in the subtalar joint Although imaging may be normal, prior to subtalar joint arthroscopy, radiographic imaging should be performed and include both of the following: Plain X-rays with one or more views (anteroposterior, lateral, axial, and/or Broden’s) to confirm and differentiate any of the following: Degenerative joint changes Loose bodies Osteochondral lesions Impingement -

In Patients with End-Stage Ankle Arthritis, How Does Total Ankle Arthroplasty Compared to Arthrodesis Affect Ankle Pain and Function?
University of the Pacific Scholarly Commons Physician's Assistant Program Capstones School of Health Sciences 4-1-2020 In Patients with End-Stage Ankle Arthritis, How Does Total Ankle Arthroplasty Compared to Arthrodesis Affect Ankle Pain and Function? Zachary Whipple University of the Pacific, [email protected] Follow this and additional works at: https://scholarlycommons.pacific.edu/pa-capstones Part of the Medicine and Health Sciences Commons Recommended Citation Whipple, Zachary, "In Patients with End-Stage Ankle Arthritis, How Does Total Ankle Arthroplasty Compared to Arthrodesis Affect Ankle Pain and Function?" (2020). Physician's Assistant Program Capstones. 84. https://scholarlycommons.pacific.edu/pa-capstones/84 This Capstone is brought to you for free and open access by the School of Health Sciences at Scholarly Commons. It has been accepted for inclusion in Physician's Assistant Program Capstones by an authorized administrator of Scholarly Commons. For more information, please contact [email protected]. In Patients with End-Stage Ankle Arthritis, How Does Total Ankle Arthroplasty Compared to Arthrodesis Affect Ankle Pain and Function? By Zachary Whipple Capstone Project Submitted to the Faculty of the Department of Physician Assistant Education of the University of the Pacific in partial fulfilment of the requirements for the degree of MASTER OF PHYSICIAN ASSISTANT STUDIES April 2020 Introduction End-stage ankle arthritis is a debilitating degenerative disease commonly located at the tibiotalar joint. The prevalence of symptomatic arthritis is about nine times lower than the rates associated with those of the knee or hip.1 Though less common than knee and hip arthritis, the US estimates greater than 50,000 new cases are reported each year.2 The most common etiology of ankle arthritis is post-traumatic pathology. -

Differences Between Subtotal Corpectomy and Laminoplasty for Cervical Spondylotic Myelopathy
Spinal Cord (2010) 48, 214–220 & 2010 International Spinal Cord Society All rights reserved 1362-4393/10 $32.00 www.nature.com/sc ORIGINAL ARTICLE Differences between subtotal corpectomy and laminoplasty for cervical spondylotic myelopathy S Shibuya1, S Komatsubara1, S Oka2, Y Kanda1, N Arima1 and T Yamamoto1 1Department of Orthopaedic Surgery, School of Medicine, Kagawa University, Kagawa, Japan and 2Oka Orthopaedic and Rehabilitation Clinic, Kagawa, Japan Objective: This study aimed to obtain guidelines for choosing between subtotal corpectomy (SC) and laminoplasty (LP) by analysing the surgical outcomes, radiological changes and problems associated with each surgical modality. Study Design: A retrospective analysis of two interventional case series. Setting: Department of Orthopaedic Surgery, Kagawa University, Japan. Methods: Subjects comprised 34 patients who underwent SC and 49 patients who underwent LP. SC was performed by high-speed drilling to remove vertebral bodies. Autologous strut bone grafting was used. LP was performed as an expansive open-door LP. The level of decompression was from C3 to C7. Clinical evaluations included recovery rate (RR), frequency of C5 root palsy after surgery, re-operation and axial pain. Radiographic assessments included sagittal cervical alignment and bone union. Results: Comparisons between the two groups showed no significant differences in age at surgery, preoperative factors, RR and frequency of C5 palsy. Progression of kyphotic changes, operation time and volumes of blood loss and blood transfusion were significantly greater in the SC (two- or three- level) group. Six patients in the SC group required additional surgery because of pseudoarthrosis, and four patients underwent re-operation because of adjacent level disc degeneration. -

Procedure Coding in ICD-9-CM and ICD- 10-PCS
Procedure Coding in ICD-9-CM and ICD- 10-PCS ICD-9-CM Volume 3 Procedures are classified in volume 3 of ICD-9-CM, and this section includes both an Alphabetic Index and a Tabular List. This volume follows the same format, organization and conventions as the classification of diseases in volumes 1 and 2. ICD-10-PCS ICD-10-PCS will replace volume 3 of ICD-9-CM. Unlike ICD-10-CM for diagnoses, which is similar in structure and format as the ICD-9-CM volumes 1 and 2, ICD-10-PCS is a completely different system. ICD-10-PCS has a multiaxial seven-character alphanumeric code structure providing unique codes for procedures. The table below gives a brief side-by-side comparison of ICD-9-CM and ICD-10-PCS. ICD-9-CM Volume3 ICD-10-PCS Follows ICD structure (designed for diagnosis Designed and developed to meet healthcare coding) needs for a procedure code system Codes available as a fixed or finite set in list form Codes constructed from flexible code components (values) using tables Codes are numeric Codes are alphanumeric Codes are 3-4 digits long All codes are seven characters long ICD-9-CM and ICD-10-PCS are used to code only hospital inpatient procedures. Hospital outpatient departments, other ambulatory facilities, and physician practices are required to use CPT and HCPCS to report procedures. ICD-9-CM Conventions in Volume 3 Code Also In volume 3, the phrase “code also” is a reminder to code additional procedures only when they have actually been performed. -

Knee Arthrodesis After Failed Total Knee Arthroplasty
650 COPYRIGHT Ó 2019 BY THE JOURNAL OF BONE AND JOINT SURGERY,INCORPORATED Current Concepts Review Knee Arthrodesis After Failed Total 04/12/2019 on 1mhtSo9F6TkBmpGAR5GLp6FT3v73JgoS8Zn360/N4fAEQXu6c15Knc+cXP2J5+wvbQY2nVcoOF2DIk3Zd0BSqmOXRD8WUDFCPOJ9CnEHMNOmtIbs3S0ykA== by http://journals.lww.com/jbjsjournal from Downloaded Knee Arthroplasty Downloaded Asim M. Makhdom, MD, MSc, FRCSC, Austin Fragomen, MD, and S. Robert Rozbruch, MD from http://journals.lww.com/jbjsjournal Investigation performed at the Hospital for Special Surgery, Weill Cornell Medicine, Cornell University, New York, NY ä Knee arthrodesis after failure of a total knee arthroplasty (TKA) because of periprosthetic joint infection (PJI) may provide superior functional outcome and ambulatory status compared with above-the-knee amputation. by 1mhtSo9F6TkBmpGAR5GLp6FT3v73JgoS8Zn360/N4fAEQXu6c15Knc+cXP2J5+wvbQY2nVcoOF2DIk3Zd0BSqmOXRD8WUDFCPOJ9CnEHMNOmtIbs3S0ykA== ä The use of an intramedullary nail (IMN) for knee arthrodesis following removal of TKA components because of a PJI may result in higher fusion rates compared with external fixation devices. ä The emerging role of the antibiotic cement-coated interlocking IMN may expand the indications to achieve knee fusion in a single-stage intervention. ä Massive bone defects after failure of an infected TKA can be managed with various surgical strategies in a single- stage intervention to preserve leg length and function. Primary total knee arthroplasty (TKA) is a common procedure suppressive antibiotics for recurrent PJIs are generally reserved with a reported increase of 162% from 1991 to 2010 in the for patients with more severe preoperative disability and United States1,2. From 2005 to 2030, it is projected that the medical comorbidity and those who are not candidates to number of TKA procedures will grow by 673% or 3.5 million. -
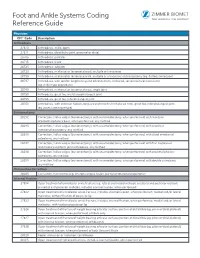
Foot and Ankle Systems Coding Reference Guide
Foot and Ankle Systems Coding Reference Guide Physician CPT® Code Description Arthrodesis 27870 Arthrodesis, ankle, open 27871 Arthrodesis, tibiofibular joint, proximal or distal 28705 Arthrodesis; pantalar 28715 Arthrodesis; triple 28725 Arthrodesis; subtalar 28730 Arthrodesis, midtarsal or tarsometatarsal, multiple or transverse 28735 Arthrodesis, midtarsal or tarsometatarsal, multiple or transverse; with osteotomy (eg, flatfoot correction) 28737 Arthrodesis, with tendon lengthening and advancement, midtarsal, tarsal navicular-cuneiform (eg, miller type procedure) 28740 Arthrodesis, midtarsal or tarsometatarsal, single joint 28750 Arthrodesis, great toe; metatarsophalangeal joint 28755 Arthrodesis, great toe; interphalangeal joint 28760 Arthrodesis, with extensor hallucis longus transfer to first metatarsal neck, great toe, interphalangeal joint (eg, jones type procedure) Bunionectomy 28292 Correction, hallux valgus (bunionectomy), with sesamoidectomy, when performed; with resection of proximal phalanx base, when performed, any method 28295 Correction, hallux valgus (bunionectomy), with sesamoidectomy, when performed; with proximal metatarsal osteotomy, any method 28296 Correction, hallux valgus (bunionectomy), with sesamoidectomy, when performed; with distal metatarsal osteotomy, any method 28297 Correction, hallux valgus (bunionectomy), with sesamoidectomy, when performed; with first metatarsal and medial cuneiform joint arthrodesis, any method 28298 Correction, hallux valgus (bunionectomy), with sesamoidectomy, when performed; with -

USE of INTERPOSITIONAL BONE GRAFTS for FIRST METARSOPHALANGEAL ARTHRODESIS: a Review of Current Literature
CHAPTER 8 USE OF INTERPOSITIONAL BONE GRAFTS FOR FIRST METARSOPHALANGEAL ARTHRODESIS: A Review of Current Literature Thomas A. Brosky, II, DPM Cayla Conway, DPM INTRODUCTION incidence of nonunion with the use of autogenic iliac crest graft (26.7%) compared to the allograft (0%). Surgical procedures for fi rst metarsophalangeal joint (MPJ) In contrast to the Myerson study, Mankovecky et al arthritis have been well outlined in podiatric literature. (2) suggested a much lower rate of nonunion with the use The fi rst MPJ fusion is a procedure that is commonly autogenous bone graft. In 2013, they reported a review of 6 used for initial treatment of this condition or subsequent studies, which reported a total of 42 procedures of fi rst MPJ treatment, after another surgical procedure has failed. A arthrodesis with autogenous graft as a salvage procedure for failed arthroplasty may result in an excessively shortened a failed Keller arthroplasty. Out of the 42 cases, there were fi rst metatarsal or an overly resected proximal phalanx, 2 reported nonunions for a nonunion rate of 4.8%, which which may necessitate the use of an interpositional bone- is signifi cantly less than the rate reported by Myerson. One block graft to restore adequate length. The preservation of of the nonunions was revised, and the other was reported length can pose a surgical challenge, however, it is integral as asymptomatic. The review states the cases were highly in preventing complications that may result from altered varied in technique and fi xation approach. Mankovecky et al biomechanics, such as inadequate propulsion and lesser reported a need for further research to standardize the data metatarsalgia. -

Knee-Arthrodesis-1.Pdf
The Knee 24 (2017) 91–99 Contents lists available at ScienceDirect The Knee Knee arthrodesis by the Ilizarov method in the treatment of total knee arthroplasty failure Andrea Antonio Maria Bruno a,⁎, Alexander Kirienko b, Andrea Peccati c, Paolo Dupplicato a, Massimo De Donato a, Enrico Arnaldi a, Nicola Portinaro c a Arthroscopic and Reconstructive Surgery of the Knee Unit, Humanitas Research Hospital, Rozzano, Milan, Italy b External Fixation Unit, Humanitas Research Hospital, Rozzano, Milan, Italy c Orthopaedics Department, University of Milan, Milan, Italy article info abstract Article history: Background: Currently, the main indication for knee arthrodesis is septic failure of a total knee Received 8 June 2015 arthroplasty (TKA). The purpose of this study was to evaluate the results of knee arthrodesis by Received in revised form 22 September 2016 circular external fixation performed in the treatment of TKA failure in which revision Accepted 3 November 2016 arthroplasty was not indicated. Methods: The study involved 19 patients who underwent knee arthrodesis by the Ilizarov method. Clinical and functional assessments were performed, including Knee Society Score Keywords: (KSS). A postoperative clinical and radiographic evaluation was conducted every three months TKA failure Ilizarov method until the end of the treatment. Postoperative complications and eventual leg shortening were Knee arthrodesis recorded. Results: KSS results showed a significant improvement with respect to the preoperative condi- tion. Of the 16 patients in the final follow-up, 15 patients (93.7%) achieved complete bone fu- sion. Major complications occurred in patients treated for septic failure of TKA and most occurred in patients over 75 years of age; the mean final leg shortening was four centimeters. -

Acknowledgement Foot & Ankle Center of Excellence • Professor Emeritus Ian J
Division of Foot and Ankle Common Foot & Reconstruction Ankle Injuries • Adam T. Groth, MD • Kevin P. Martin, DO • Timothy Miller, MD Adam T. Groth, MD • Julie Swain, APRN-CNP Associate Professor Chief, Division of Foot & Ankle Reconstruction • OSU Podiatry Jameson Crane Sports Medicine Institute Department of Orthopaedics • OSU Sports Medicine Family Medicine The Ohio State University Wexner Medical Center • OSU PM&R • OSU Physical Therapy Acknowledgement Foot & Ankle Center of Excellence • Professor Emeritus Ian J. Alexander • Comprehensive care for all adult foot and ankle Disclosures problems: • None • Sports injuries / Sprains / Cartilage disorders • Arthritis / Degenerative conditions • Deformities • Trauma / Fractures • Bunions / Hammertoes • Whatever is causing your pain 1 Common Problems of the Acute Ankle Sprain Foot & Ankle • Exceedingly common • Acute ankle sprains • 10-40% of civilian athletic injuries annually • Significant time lost to injury • Late pain after ankle sprains / associated injuries • 1 inversion event per 10,000 people per day • Stress fractures • 23,000 to 30,000 ankle injuries per day in U.S. • 10% or ER visits in U.S. • Achilles tendon ruptures • Plantar fasciitis • 45% of all basketball injuries • Bunions • 31% of collegiate football injuries • 20% of soccer injuries • Ankle arthritis • Leading cause of time loss in NFL • Most common cause of acute injury in volleyball The Ankle Sprain Anatomy and Biomechanics • Mainstay of treatment is functional rehabilitation • 80% make a full recovery with conservative -

Cervical Laminoplasty Michael P
The Spine Journal 6 (2006) 274S–281S Cervical laminoplasty Michael P. Steinmetz, MD, Daniel K. Resnick, MD* Department of Neurosurgery, University of Wisconsin School of Medicine, K4/834 Clinical Science Center, 600 Highland Ave., Madison, WI 53792, USA Abstract Laminoplasty was developed to treat multilevel pathology of the cervical spine, namely ossification of the posterior longitudinal ligament and cervical spondylotic myelopathy. Laminoplasty was pop- ularized in the 1980s, and since then many variations on the theme have been developed. All are similar in that they expand the cervical canal while leaving the protective dorsal elements in place. Advocates claim that this prevents the formation of the ‘‘postlaminectomy’’ membrane, maintains spinal alignment, and should aid in maintaining cervical range of motion. The aforementioned are all potential shortcomings of laminectomy or laminectomy and fusion. The procedure has proven to be essentially equal to other cervical decompressive procedures in the neutral or lordotic spine, and outcome has been shown to be durable. Ó 2006 Elsevier Inc. All rights reserved. Keywords: Laminoplasty; Cervical spine; Laminectomy; Ossification of the posterior longitudinal ligament; Cervical spon- dylotic myelopathy; Myelopathy; Cervical stenosis Introduction potentially reduce the incidence of postoperative instability. Cervical motion is theoretically preserved. In 1973, Oyama Cervical laminectomy has long been the treatment for et al. introduced a Z-plasty of the cervical spine [1]. This multilevel cervical spondylosis. It permits adequate decom- procedure allowed decompression, while the retained lam- pression of the cervical spinal cord and is safe and easily inae provided support and prevented invasion of the postla- performed. Potential adverse outcomes after cervical lami- minectomy membrane. -

Development of the ICD-10 Procedure Coding System (ICD-10-PCS)
Development of the ICD-10 Procedure Coding System (ICD-10-PCS) Richard F. Averill, M.S., Robert L. Mullin, M.D., Barbara A. Steinbeck, RHIT, Norbert I. Goldfield, M.D, Thelma M. Grant, RHIA, Rhonda R. Butler, CCS, CCS-P The International Classification of Diseases 10th Revision Procedure Coding System (ICD-10-PCS) has been developed as a replacement for Volume 3 of the International Classification of Diseases 9th Revision (ICD-9-CM). The development of ICD-10-PCS was funded by the U.S. Centers for Medicare and Medicaid Services (CMS).1 ICD-10- PCS has a multiaxial seven character alphanumeric code structure that provides a unique code for all substantially different procedures, and allows new procedures to be easily incorporated as new codes. ICD10-PCS was under development for over five years. The initial draft was formally tested and evaluated by an independent contractor; the final version was released in the Spring of 1998, with annual updates since the final release. The design, development and testing of ICD-10-PCS are discussed. Introduction Volume 3 of the International Classification of Diseases 9th Revision Clinical Modification (ICD-9-CM) has been used in the U.S. for the reporting of inpatient pro- cedures since 1979. The structure of Volume 3 of ICD-9-CM has not allowed new procedures associated with rapidly changing technology to be effectively incorporated as new codes. As a result, in 1992 the U.S. Centers for Medicare and Medicaid Services (CMS) funded a project to design a replacement for Volume 3 of ICD-9-CM. -

7.01.550 Knee Arthroplasty in Adults
MEDICAL POLICY – 7.01.550 Knee Arthroplasty in Adults Effective Date: Oct. 1, 2021 RELATED MEDICAL POLICIES: Last Revised: Sept. 2, 2021 7.01.15 Meniscal Allograft and Other Meniscal Implants Replaces: N/A 7.01.144 Patient-Specific Cutting Guides for Joint Arthroplasty 7.01.549 Knee Arthroscopy in Adults 7.01.568 Designated Centers of Excellence: Total Knee or Total Hip Replacement 7.01.573 Hip Arthroplasty in Adults Select a hyperlink below to be directed to that section. POLICY CRITERIA | DOCUMENTATION REQUIREMENTS | CODING RELATED INFORMATION | EVIDENCE REVIEW | REFERENCES | HISTORY ∞ Clicking this icon returns you to the hyperlinks menu above. Introduction Knee arthroplasty is the medical term for a total knee replacement. A surgeon removes the damaged part of the joint. The surfaces are shaped to hold a replacement joint that is either metal or plastic. The artificial joint is attached to the thigh bone, shin bone, and knee cap. For the right patient, a knee replacement reduces pain and improves quality of life. People who may qualify for this surgery are those who have severe pain from “wear-and-tear” arthritis (osteoarthritis) of the knee, who are not able to perform their normal daily activities, and who tried and failed nonsurgical treatments. Replacement joints have a limited life. Factors such as a person’s age, severity of knee disease, obesity, and the type of replacement affect how long an artificial joint will last. Knee arthroplasty must be pre-approved by the health plan. This policy outlines the information needed for health plan review. Note: The Introduction section is for your general knowledge and is not to be taken as policy coverage criteria.