Pulmonary Fibrosis Associated with TINF2 Gene Mutation: Is Somatic Reversion Required?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC)
cancers Review Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC) Chiara Diquigiovanni * and Elena Bonora Unit of Medical Genetics, Department of Medical and Surgical Sciences, University of Bologna, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-208-8418 Simple Summary: Non-medullary thyroid carcinoma (NMTC) originates from thyroid follicular epithelial cells and is considered familial when occurs in two or more first-degree relatives of the patient, in the absence of predisposing environmental factors. Familial NMTC (FNMTC) cases show a high genetic heterogeneity, thus impairing the identification of pivotal molecular changes. In the past years, linkage-based approaches identified several susceptibility loci and variants associated with NMTC risk, however only few genes have been identified. The advent of next-generation sequencing technologies has improved the discovery of new predisposing genes. In this review we report the most significant genes where variants predispose to FNMTC, with the perspective that the integration of these new molecular findings in the clinical data of patients might allow an early detection and tailored therapy of the disease, optimizing patient management. Abstract: Non-medullary thyroid carcinoma (NMTC) is the most frequent endocrine tumor and originates from the follicular epithelial cells of the thyroid. Familial NMTC (FNMTC) has been defined in pedigrees where two or more first-degree relatives of the patient present the disease in absence of other predisposing environmental factors. Compared to sporadic cases, FNMTCs are often multifocal, recurring more frequently and showing an early age at onset with a worse outcome. FNMTC cases Citation: Diquigiovanni, C.; Bonora, E. -

The Genetics and Clinical Manifestations of Telomere Biology Disorders Sharon A
REVIEW The genetics and clinical manifestations of telomere biology disorders Sharon A. Savage, MD1, and Alison A. Bertuch, MD, PhD2 3 Abstract: Telomere biology disorders are a complex set of illnesses meric sequence is lost with each round of DNA replication. defined by the presence of very short telomeres. Individuals with classic Consequently, telomeres shorten with aging. In peripheral dyskeratosis congenita have the most severe phenotype, characterized blood leukocytes, the cells most extensively studied, the rate 4 by the triad of nail dystrophy, abnormal skin pigmentation, and oral of attrition is greatest during the first year of life. Thereafter, leukoplakia. More significantly, these individuals are at very high risk telomeres shorten more gradually. When the extent of telo- of bone marrow failure, cancer, and pulmonary fibrosis. A mutation in meric DNA loss exceeds a critical threshold, a robust anti- one of six different telomere biology genes can be identified in 50–60% proliferative signal is triggered, leading to cellular senes- of these individuals. DKC1, TERC, TERT, NOP10, and NHP2 encode cence or apoptosis. Thus, telomere attrition is thought to 1 components of telomerase or a telomerase-associated factor and TINF2, contribute to aging phenotypes. 5 a telomeric protein. Progressively shorter telomeres are inherited from With the 1985 discovery of telomerase, the enzyme that ex- generation to generation in autosomal dominant dyskeratosis congenita, tends telomeric nucleotide repeats, there has been rapid progress resulting in disease anticipation. Up to 10% of individuals with apparently both in our understanding of basic telomere biology and the con- acquired aplastic anemia or idiopathic pulmonary fibrosis also have short nection of telomere biology to human disease. -

Structural and Functional Consequences of a Disease Mutation in the Telomere Protein TPP1
Structural and functional consequences of a disease mutation in the telomere protein TPP1 Kamlesh Bishta,1, Eric M. Smitha,b,1, Valerie M. Tesmera, and Jayakrishnan Nandakumara,b,2 aDepartment of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109; and bProgram in Chemical Biology, University of Michigan, Ann Arbor, MI 48109 Edited by Joachim Lingner, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland, and accepted by Editorial Board Member Dinshaw J. Patel September 29, 2016 (received for review April 8, 2016) Telomerase replicates chromosome ends to facilitate continued cell The discovery of the TEL patch of TPP1 prompted the pre- division. Mutations that compromise telomerase function result in diction that this region could be a hotspot for mutations that stem cell failure diseases, such as dyskeratosis congenita (DC). One cause telomerase-deficiency diseases, such as DC. We recently such mutation (K170Δ), residing in the telomerase-recruitment factor reported a case of a severe variant of DC, Hoyeraal–Hreidarsson TPP1, provides an excellent opportunity to structurally, biochemi- syndrome (23), in which the proband was heterozygous for a cally, and genetically dissect the mechanism of such diseases. We deletion of a single amino acid of the TPP1 protein, namely lysine ACD show through site-directed mutagenesis and X-ray crystallography 170 (K170) (24). Our study placed the gene coding for TPP1 DKC1 TERC TERT that this TPP1 disease mutation deforms the conformation of two protein on a list with 10 other genes ( , , , RTEL1 TINF2 CTC1 NOP10 NHP2 WRAP53 PARN critical amino acids of the TEL [TPP1’s glutamate (E) and leucine-rich , , , , , ,and ) that are found mutated in DC and other telomere-related dis- (L)] patch, the surface of TPP1 that binds telomerase. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -

Structural and Functional Consequences of a Disease Mutation in the Telomere Protein TPP1
Structural and functional consequences of a disease mutation in the telomere protein TPP1 Kamlesh Bishta,1, Eric M. Smitha,b,1, Valerie M. Tesmera, and Jayakrishnan Nandakumara,b,2 aDepartment of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109; and bProgram in Chemical Biology, University of Michigan, Ann Arbor, MI 48109 Edited by Joachim Lingner, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland, and accepted by Editorial Board Member Dinshaw J. Patel September 29, 2016 (received for review April 8, 2016) Telomerase replicates chromosome ends to facilitate continued cell The discovery of the TEL patch of TPP1 prompted the pre- division. Mutations that compromise telomerase function result in diction that this region could be a hotspot for mutations that stem cell failure diseases, such as dyskeratosis congenita (DC). One cause telomerase-deficiency diseases, such as DC. We recently such mutation (K170Δ), residing in the telomerase-recruitment factor reported a case of a severe variant of DC, Hoyeraal–Hreidarsson TPP1, provides an excellent opportunity to structurally, biochemi- syndrome (23), in which the proband was heterozygous for a cally, and genetically dissect the mechanism of such diseases. We deletion of a single amino acid of the TPP1 protein, namely lysine ACD show through site-directed mutagenesis and X-ray crystallography 170 (K170) (24). Our study placed the gene coding for TPP1 DKC1 TERC TERT that this TPP1 disease mutation deforms the conformation of two protein on a list with 10 other genes ( , , , RTEL1 TINF2 CTC1 NOP10 NHP2 WRAP53 PARN critical amino acids of the TEL [TPP1’s glutamate (E) and leucine-rich , , , , , ,and ) that are found mutated in DC and other telomere-related dis- (L)] patch, the surface of TPP1 that binds telomerase. -

A Role for Heterochromatin Protein 1G at Human Telomeres
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press A role for heterochromatin protein 1g at human telomeres Silvia Canudas,1 Benjamin R. Houghtaling,1 Monica Bhanot,1 Ghadir Sasa,2 Sharon A. Savage,3 Alison A. Bertuch,2,4 and Susan Smith1,5 1Molecular Pathogenesis Program, Department of Pathology, Kimmel Center for Biology and Medicine of the Skirball Institute, New York University School of Medicine, New York, New York 10016, USA; 2Department of Pediatrics, Baylor College of Medicine, Houston, Texas 77030, USA; 3Clinical Genetics Branch Division of Cancer Epidemiology and Genetics, National Cancer Institute, Rockville, Maryland 20892, USA; 4Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA Human telomere function is mediated by shelterin, a six-subunit complex that is required for telomere replication, protection, and cohesion. TIN2, the central component of shelterin, has binding sites to three subunits: TRF1, TRF2, and TPP1. Here we identify a fourth partner, heterochromatin protein 1g (HP1g), that binds to a conserved canonical HP1-binding motif, PXVXL, in the C-terminal domain of TIN2. We show that HP1g localizes to telomeres in S phase, where it is required to establish/maintain cohesion. We further demonstrate that the HP1- binding site in TIN2 is required for sister telomere cohesion and can impact telomere length maintenance by telomerase. Remarkably, the PTVML HP1-binding site is embedded in the recently identified cluster of mutations in TIN2 that gives rise to dyskeratosis congenita (DC), an inherited bone marrow failure syndrome caused by defects in telomere maintenance. -

Engineered Type 1 Regulatory T Cells Designed for Clinical Use Kill Primary
ARTICLE Acute Myeloid Leukemia Engineered type 1 regulatory T cells designed Ferrata Storti Foundation for clinical use kill primary pediatric acute myeloid leukemia cells Brandon Cieniewicz,1* Molly Javier Uyeda,1,2* Ping (Pauline) Chen,1 Ece Canan Sayitoglu,1 Jeffrey Mao-Hwa Liu,1 Grazia Andolfi,3 Katharine Greenthal,1 Alice Bertaina,1,4 Silvia Gregori,3 Rosa Bacchetta,1,4 Norman James Lacayo,1 Alma-Martina Cepika1,4# and Maria Grazia Roncarolo1,2,4# Haematologica 2021 Volume 106(10):2588-2597 1Department of Pediatrics, Division of Stem Cell Transplantation and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 2Stanford Institute for Stem Cell Biology and Regenerative Medicine, Stanford School of Medicine, Stanford, CA, USA; 3San Raffaele Telethon Institute for Gene Therapy, Milan, Italy and 4Center for Definitive and Curative Medicine, Stanford School of Medicine, Stanford, CA, USA *BC and MJU contributed equally as co-first authors #AMC and MGR contributed equally as co-senior authors ABSTRACT ype 1 regulatory (Tr1) T cells induced by enforced expression of interleukin-10 (LV-10) are being developed as a novel treatment for Tchemotherapy-resistant myeloid leukemias. In vivo, LV-10 cells do not cause graft-versus-host disease while mediating graft-versus-leukemia effect against adult acute myeloid leukemia (AML). Since pediatric AML (pAML) and adult AML are different on a genetic and epigenetic level, we investigate herein whether LV-10 cells also efficiently kill pAML cells. We show that the majority of primary pAML are killed by LV-10 cells, with different levels of sensitivity to killing. Transcriptionally, pAML sensitive to LV-10 killing expressed a myeloid maturation signature. -

The Shelterin Complex and Hematopoiesis
The shelterin complex and hematopoiesis Morgan Jones, … , Catherine E. Keegan, Ivan Maillard J Clin Invest. 2016;126(5):1621-1629. https://doi.org/10.1172/JCI84547. Review Mammalian chromosomes terminate in stretches of repetitive telomeric DNA that act as buffers to avoid loss of essential genetic information during end-replication. A multiprotein complex known as shelterin prevents recognition of telomeric sequences as sites of DNA damage. Telomere erosion contributes to human diseases ranging from BM failure to premature aging syndromes and cancer. The role of shelterin telomere protection is less understood. Mutations in genes encoding the shelterin proteins TRF1-interacting nuclear factor 2 (TIN2) and adrenocortical dysplasia homolog (ACD) were identified in dyskeratosis congenita, a syndrome characterized by somatic stem cell dysfunction in multiple organs leading to BM failure and other pleiotropic manifestations. Here, we introduce the biochemical features and in vivo effects of individual shelterin proteins, discuss shelterin functions in hematopoiesis, and review emerging knowledge implicating the shelterin complex in hematological disorders. Find the latest version: https://jci.me/84547/pdf The Journal of Clinical Investigation REVIEW The shelterin complex and hematopoiesis Morgan Jones,1,2 Kamlesh Bisht,3 Sharon A. Savage,4 Jayakrishnan Nandakumar,3 Catherine E. Keegan,5,6 and Ivan Maillard7,8,9 1Graduate Program in Cellular and Molecular Biology, 2Medical Scientist Training Program, and 3Department of Molecular, Cellular and Developmental Biology, University of Michigan, Ann Arbor, Michigan, USA. 4Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, Bethesda, Maryland, USA. 5Department of Human Genetics, 6Department of Pediatrics, 7Life Sciences Institute, 8Division of Hematology-Oncology, Department of Internal Medicine, and 9Department of Cell and Developmental Biology, University of Michigan, Ann Arbor, Michigan, USA. -
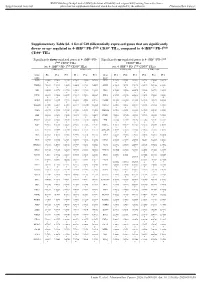
Supplementary Table S6. a List of 520 Differentially Expressed Genes
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S6. A list of 520 differentially expressed genes that are significantly down- or up- regulated in 4-1BBpos PD-1high CD39+ TILs, compared to 4-1BBneg PD-1high CD39+ TILs Significantly down-regulated genes in 4-1BBpos PD- Significantly up-regulated genes in 4-1BBpos PD-1high 1high CD39+ TILs CD39+ TILs (vs. 4-1BBneg PD-1high CD39+ TILs) (vs. 4-1BBneg PD-1high CD39+ TILs) 4-1BBneg cells 4-1BBpos cells 4-1BBneg cells 4-1BBpos cells Gene Pt 1 Pt 2 Pt 3 Pt 1 Pt 2 Pt 3 Gene Pt 1 Pt 2 Pt 3 Pt 1 Pt 2 Pt 3 name name FOSB 9.92496 9.80436 9.61180 9.76085 9.55460 8.47625 SPC25 2.82975 2.53201 1.83699 3.05751 3.79646 3.66172 5 9 4 4 3 9 8 5 5 6 7 PTGER4 7.26693 7.28598 7.19060 6.84482 6.61319 5.90088 GMNN 4.55416 5.05351 3.79176 4.95671 5.63164 4.93921 3 5 1 4 8 7 8 5 3 3 ABI3 6.24122 6.45779 6.27735 5.85419 5.81360 5.18958 ING3 4.78602 5.23034 4.49070 5.22964 5.62759 5.34212 1 3 5 6 6 2 3 6 5 8 7 1 LITAF 9.06689 9.17420 8.99478 8.79217 8.74965 8.08497 KIF14 4.58732 4.59859 4.62494 5.12658 5.70643 5.26241 4 4 9 8 7 5 7 9 3 7 4 ZNF10 4.90912 4.98940 4.74775 4.33282 4.40018 3.50431 CENPE 6.54529 6.55450 6.47696 6.84945 7.22848 6.96595 8 7 8 8 2 8 5 7 9 5 3 2 TNFAIP3 11.4201 11.2484 11.4068 11.1877 10.9900 10.5350 CDCA7 3.80074 4.20586 3.92787 4.43496 5.46931 4.40814 9 4 2 9 7 9 9 5 8 NAPA 5.94770 5.79864 5.64849 -

Cell Culture-Based Profiling Across Mammals Reveals DNA Repair And
1 Cell culture-based profiling across mammals reveals 2 DNA repair and metabolism as determinants of 3 species longevity 4 5 Siming Ma1, Akhil Upneja1, Andrzej Galecki2,3, Yi-Miau Tsai2, Charles F. Burant4, Sasha 6 Raskind4, Quanwei Zhang5, Zhengdong D. Zhang5, Andrei Seluanov6, Vera Gorbunova6, 7 Clary B. Clish7, Richard A. Miller2, Vadim N. Gladyshev1* 8 9 1 Division of Genetics, Department of Medicine, Brigham and Women’s Hospital, Harvard 10 Medical School, Boston, MA, 02115, USA 11 2 Department of Pathology and Geriatrics Center, University of Michigan Medical School, 12 Ann Arbor, MI 48109, USA 13 3 Department of Biostatistics, School of Public Health, University of Michigan, Ann Arbor, 14 MI 48109, USA 15 4 Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI 16 48109, USA 17 5 Department of Genetics, Albert Einstein College of Medicine, Bronx, NY 10128, USA 18 6 Department of Biology, University of Rochester, Rochester, NY 14627, USA 19 7 Broad Institute, Cambridge, MA 02142, US 20 21 * corresponding author: Vadim N. Gladyshev ([email protected]) 22 ABSTRACT 23 Mammalian lifespan differs by >100-fold, but the mechanisms associated with such 24 longevity differences are not understood. Here, we conducted a study on primary skin 25 fibroblasts isolated from 16 species of mammals and maintained under identical cell culture 26 conditions. We developed a pipeline for obtaining species-specific ortholog sequences, 27 profiled gene expression by RNA-seq and small molecules by metabolite profiling, and 28 identified genes and metabolites correlating with species longevity. Cells from longer-lived 29 species up-regulated genes involved in DNA repair and glucose metabolism, down-regulated 30 proteolysis and protein transport, and showed high levels of amino acids but low levels of 31 lysophosphatidylcholine and lysophosphatidylethanolamine.