Meta-Analysis of Differentially Expressed Genes in Autism Based on Gene Expression Data
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

High-Throughput Bioinformatics Approaches to Understand Gene Expression Regulation in Head and Neck Tumors
High-throughput bioinformatics approaches to understand gene expression regulation in head and neck tumors by Yanxiao Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in The University of Michigan 2016 Doctoral Committee: Associate Professor Maureen A. Sartor, Chair Professor Thomas E. Carey Assistant Professor Hui Jiang Professor Ronald J. Koenig Associate Professor Laura M. Rozek Professor Kerby A. Shedden c Yanxiao Zhang 2016 All Rights Reserved I dedicate this thesis to my family. For their unfailing love, understanding and support. ii ACKNOWLEDGEMENTS I would like to express my gratitude to Dr. Maureen Sartor for her guidance in my research and career development. She is a great mentor. She patiently taught me when I started new in this field, granted me freedom to explore and helped me out when I got lost. Her dedication to work, enthusiasm in teaching, mentoring and communicating science have inspired me to feel the excite- ment of research beyond novel scientific discoveries. I’m also grateful to have an interdisciplinary committee. Their feedback on my research progress and presentation skills is very valuable. In particular, I would like to thank Dr. Thomas Carey and Dr. Laura Rozek for insightful discussions on the biology of head and neck cancers and human papillomavirus, Dr. Ronald Koenig for expert knowledge on thyroid cancers, Dr. Hui Jiang and Dr. Kerby Shedden for feedback on the statistics part of my thesis. I would like to thank all the past and current members of Sartor lab for making the lab such a lovely place to stay and work in. -

Biallelic Mutations in PLA2G5, Encoding Group V Phospholipase A2, Cause Benign Fleck Retina
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector REPORT Biallelic Mutations in PLA2G5, Encoding Group V Phospholipase A2, Cause Benign Fleck Retina Panagiotis I. Sergouniotis,1,2,7 Alice E. Davidson,1,7 Donna S. Mackay,1 Eva Lenassi,1,2 Zheng Li,1,8 Anthony G. Robson,1,2 Xu Yang,3 Jaimie Hoh Kam,1 Timothy W. Isaacs,4 Graham E. Holder,1,2 Glen Jeffery,1 Jonathan A. Beck,5 Anthony T. Moore,1,2 Vincent Plagnol,6 and Andrew R. Webster1,2,* Flecked-retina syndromes, including fundus flavimaculatus, fundus albipunctatus, and benign fleck retina, comprise a group of disorders with widespread or limited distribution of yellow-white retinal lesions of various sizes and configurations. Three siblings who have benign fleck retina and were born to consanguineous parents are the basis of this report. A combination of homozygosity mapping and exome sequencing helped to identify a homozygous missense mutation, c.133G>T (p.Gly45Cys), in PLA2G5, a gene encoding a secreted phospholipase (group V phospholipase A2). A screen of a further four unrelated individuals with benign fleck retina detected biallelic variants in the same gene in three patients. In contrast, no loss of function or common (minor-allele frequency>0.05%) nonsynonymous PLA2G5 variants have been previously reported (EVS, dbSNP, 1000 Genomes Project) or were detected in an internal database of 224 exomes (from subjects with adult onset neurodegenerative disease and without a diagnosis of ophthalmic disease). All seven affected individuals had fundoscopic features compatible with those previously described in benign fleck retina and no visual or electrophysiological deficits. -

Reveals of Candidate Active Ingredients in Justicia and Its Anti-Thrombotic Action of Mechanism Based on Network Pharmacology Ap
www.nature.com/scientificreports OPEN Reveals of candidate active ingredients in Justicia and its anti‑thrombotic action of mechanism based on network pharmacology approach and experimental validation Zongchao Hong1,5, Ting Zhang2,5, Ying Zhang1, Zhoutao Xie1, Yi Lu1, Yunfeng Yao1, Yanfang Yang1,3,4*, Hezhen Wu1,3,4* & Bo Liu1,3* Thrombotic diseases seriously threaten human life. Justicia, as a common Chinese medicine, is usually used for anti‑infammatory treatment, and further studies have found that it has an inhibitory efect on platelet aggregation. Therefore, it can be inferred that Justicia can be used as a therapeutic drug for thrombosis. This work aims to reveal the pharmacological mechanism of the anti‑thrombotic efect of Justicia through network pharmacology combined with wet experimental verifcation. During the analysis, 461 compound targets were predicted from various databases and 881 thrombus‑related targets were collected. Then, herb‑compound‑target network and protein–protein interaction network of disease and prediction targets were constructed and cluster analysis was applied to further explore the connection between the targets. In addition, Gene Ontology (GO) and pathway (KEGG) enrichment were used to further determine the association between target proteins and diseases. Finally, the expression of hub target proteins of the core component and the anti‑thrombotic efect of Justicia’s core compounds were verifed by experiments. In conclusion, the core bioactive components, especially justicidin D, can reduce thrombosis by regulating F2, MMP9, CXCL12, MET, RAC1, PDE5A, and ABCB1. The combination of network pharmacology and the experimental research strategies proposed in this paper provides a comprehensive method for systematically exploring the therapeutic mechanism of multi‑component medicine. -

Piperlongumine) in Cancer Tiago Henrique1,10, Caroline De F
www.nature.com/scientificreports OPEN Biological and physical approaches on the role of piplartine (piperlongumine) in cancer Tiago Henrique1,10, Caroline de F. Zanon2,10, Ana P. Girol2,3, Ana Carolina Buzzo Stefanini1,9, Nayara S. de A. Contessoto4, Nelson J. F. da Silveira5, Daniel P. Bezerra6, Edilberto R. Silveira7, José M. Barbosa‑Filho8, Marinonio L. Cornélio4, Sonia M. Oliani2 & Eloiza H. Tajara1,9* Chronic infammation provides a favorable microenvironment for tumorigenesis, which opens opportunities for targeting cancer development and progression. Piplartine (PL) is a biologically active alkaloid from long peppers that exhibits anti‑infammatory and antitumor activity. In the present study, we investigated the physical and chemical interactions of PL with anti‑infammatory compounds and their efects on cell proliferation and migration and on the gene expression of infammatory mediators. Molecular docking data and physicochemical analysis suggested that PL shows potential interactions with a peptide of annexin A1 (ANXA1), an endogenous anti‑infammatory mediator with therapeutic potential in cancer. Treatment of neoplastic cells with PL alone or with annexin A1 mimic peptide reduced cell proliferation and viability and modulated the expression of MCP‑1 chemokine, IL‑8 cytokine and genes involved in infammatory processes. The results also suggested an inhibitory efect of PL on tubulin expression. In addition, PL apparently had no infuence on cell migration and invasion at the concentration tested. Considering the role of infammation in the context of promoting tumor initiation, the present study shows the potential of piplartine as a therapeutic immunomodulator for cancer prevention and progression. Several epidemiological studies have indicated that persistent infection and chronic infammation are predis- posing factors for cancer, which is well-documented for the cervix afer HPV infection1 and the stomach in the presence of Helicobacter pylori2. -

Immune Cells in Spleen and Mucosa + CD127 Neg Production of IL-17
Downloaded from http://www.jimmunol.org/ by guest on September 26, 2021 neg is online at: average * The Journal of Immunology published online 21 June 2010 Immune Cells in Spleen and Mucosa from submission to initial decision + 4 weeks from acceptance to publication TLR5 Signaling Stimulates the Innate Production of IL-17 and IL-22 by CD3 CD127 Laurye Van Maele, Christophe Carnoy, Delphine Cayet, Pascal Songhet, Laure Dumoutier, Isabel Ferrero, Laure Janot, François Erard, Julie Bertout, Hélène Leger, Florent Sebbane, Arndt Benecke, Jean-Christophe Renauld, Wolf-Dietrich Hardt, Bernhard Ryffel and Jean-Claude Sirard http://www.jimmunol.org/content/early/2010/06/21/jimmun ol.1000115 J Immunol Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2010/06/21/jimmunol.100011 5.DC1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 26, 2021. Published June 21, 2010, doi:10.4049/jimmunol.1000115 -

Rnaseq Studies Reveal Distinct Transcriptional Response to Vitamin
bioRxiv preprint doi: https://doi.org/10.1101/798504; this version posted May 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 RNAseq studies reveal distinct transcriptional response to vitamin 2 A deficiency in small intestine versus colon, uncovering novel 3 vitamin A-regulated genes 4 5 Zhi Chai1,2,*, Yafei Lyu3,8, Qiuyan Chen2, Cheng-Hsin Wei2,9, Lindsay M. Snyder4,10, Veronika 6 Weaver4, Aswathy Sebastian5, István Albert6, Qunhua Li7, Margherita T. Cantorna4, A. Catharine 2* 7 Ross . 8 9 1Intercollege Graduate Degree Program in Physiology, 2Department of Nutritional Sciences, 10 3Intercollege Graduate Degree Program in Bioinformatics and Genomics, 4Department of 11 Veterinary and Biomedical Sciences, 5Huck Institutes of the Life Sciences, 6Department of 12 Biochemistry and Molecular Biology, 7Department of Statistics. The Pennsylvania State 13 University, University Park, PA, USA. 8Present address: Department of Biostatistics, 14 Epidemiology and Informatics, Perelman School of Medicine, University of Pennsylvania, 15 Philadelphia, PA, USA. 9Present address: Frederick National Laboratory for Cancer Research, 16 Frederick, MD, USA. 10Present address: Center for Evolutionary and Theoretical Immunology, 17 The University of New Mexico, Albuquerque, NM, USA. 18 19 *Corresponding authors 20 A. Catharine Ross ([email protected]) and Zhi Chai ([email protected]) 21 110 Chandlee Laboratory 22 University Park, PA, USA. 16802 bioRxiv preprint doi: https://doi.org/10.1101/798504; this version posted May 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Pulmonary Expression of Pla2g5 During Lung Damage in Mice Induced by Fipronil and Lipopolysaccharide Interaction
Journal of Applied and Natural Science 11(2): 285- 290 (2019) ISSN : 0974-9411 (Print), 2231-5209 (Online) journals.ansfoundation.org Pulmonary expression of Pla2g5 during lung damage in mice induced by fipronil and lipopolysaccharide interaction Arif Ahmad Pandit * Article Info Department of Animal Biotechnology, School of Animal Biotechnology, Guru Angad Dev DOI: 10.31018/jans.v11i2.2043 Veterinary and Animals Sciences University, Ludhiana-141004 (Punjab), India Received: March 19, 2019 R.S. Sethi Revised: April 8, 2019 Department of Animal Biotechnology, School of Animal Biotechnology, Guru Angad Dev Accepted: April 17, 2019 Veterinary and Animals Sciences University, Ludhiana-141004 (Punjab), India *Corresponding author. E-mail: [email protected] How to Cite Abstract Pandit, A. A. and Sethi, With Indian pesticide industry touching new heights each year in terms of production and R.S. (2019). Pulmonary sale, its ill effects on health cannot just be ignored. Humans, as well as animals, are un- expression of Pla2g5 der the constant threat of being exposed to environmental pollutants like endotoxins and during lung damage in pesticides which are present ubiquitously in our surroundings. Phenylpyrazoles like mice induced by fipronil fipronil were introduced into the Indian market with an aim to oversee the menace of in- and lipopolysaccharide secticide resistance and public health hazards experienced with more commonly used interaction. Journal of Ap- pesticide but its adverse effects on the pulmonary system are now being reported global- plied and Natural Science, ly. We reported first data that the Planar cell Polarity (PCP) pathway was the top dysregu- 11(2): 285- 290 https:// lated pathway during fipronil induced lung damage. -

Updating Phospholipase A2 Biology
biomolecules Review Updating Phospholipase A2 Biology Makoto Murakami * , Hiroyasu Sato and Yoshitaka Taketomi Laboratory of Microenvironmental and Metabolic Health Sciences, Center for Disease Biology and Integrative Medicine, Graduate School of Medicine, The University of Tokyo, Bunkyo-ku, Tokyo 113-8655, Japan; [email protected] (H.S.); [email protected] (Y.T.) * Correspondence: [email protected]; Tel.: +81-3-5841-1431 Received: 24 September 2020; Accepted: 15 October 2020; Published: 19 October 2020 Abstract: The phospholipase A2 (PLA2) superfamily contains more than 50 enzymes in mammals that are subdivided into several distinct families on a structural and biochemical basis. In principle, PLA2 has the capacity to hydrolyze the sn-2 position of glycerophospholipids to release fatty acids and lysophospholipids, yet several enzymes in this superfamily catalyze other reactions rather than or in addition to the PLA2 reaction. PLA2 enzymes play crucial roles in not only the production of lipid mediators, but also membrane remodeling, bioenergetics, and body surface barrier, thereby participating in a number of biological events. Accordingly, disturbance of PLA2-regulated lipid metabolism is often associated with various diseases. This review updates the current state of understanding of the classification, enzymatic properties, and biological functions of various enzymes belonging to the PLA2 superfamily, focusing particularly on the novel roles of PLA2s in vivo. Keywords: fatty acid; knockout mouse; lipid mediator; lipidomics; lysophospholipid; membrane; phospholipase A2; phospholipid 1. Introduction Based on their structural relationships, the PLA2 superfamily is classified into several 2+ families, including the secreted PLA2 (sPLA2), cytosolic PLA2 (cPLA2), Ca -independent PLA2 (iPLA2, also called patatin-like phospholipase (PNPLA)), platelet-activating factor acetylhydrolase (PAF-AH), lysosomal PLA2 (LPLA2), PLA/acyltransferase (PLAAT), α/β hydrolase (ABHD), and glycosylphosphatidylinositol (GPI)-specific PLA2 families. -

Data-Driven and Knowledge-Driven Computational Models of Angiogenesis in Application to Peripheral Arterial Disease
DATA-DRIVEN AND KNOWLEDGE-DRIVEN COMPUTATIONAL MODELS OF ANGIOGENESIS IN APPLICATION TO PERIPHERAL ARTERIAL DISEASE by Liang-Hui Chu A dissertation submitted to Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland March, 2015 © 2015 Liang-Hui Chu All Rights Reserved Abstract Angiogenesis, the formation of new blood vessels from pre-existing vessels, is involved in both physiological conditions (e.g. development, wound healing and exercise) and diseases (e.g. cancer, age-related macular degeneration, and ischemic diseases such as coronary artery disease and peripheral arterial disease). Peripheral arterial disease (PAD) affects approximately 8 to 12 million people in United States, especially those over the age of 50 and its prevalence is now comparable to that of coronary artery disease. To date, all clinical trials that includes stimulation of VEGF (vascular endothelial growth factor) and FGF (fibroblast growth factor) have failed. There is an unmet need to find novel genes and drug targets and predict potential therapeutics in PAD. We use the data-driven bioinformatic approach to identify angiogenesis-associated genes and predict new targets and repositioned drugs in PAD. We also formulate a mechanistic three- compartment model that includes the anti-angiogenic isoform VEGF165b. The thesis can serve as a framework for computational and experimental validations of novel drug targets and drugs in PAD. ii Acknowledgements I appreciate my advisor Dr. Aleksander S. Popel to guide my PhD studies for the five years at Johns Hopkins University. I also appreciate several professors on my thesis committee, Dr. Joel S. Bader, Dr. -
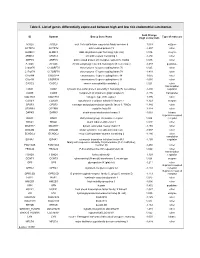
Table II. List of Genes Differentially Expressed Between High and Low Risk Endometrial Carcinomas
Table II. List of genes differentially expressed between high and low risk endometrial carcinomas Fold Change ID Symbol Entrez Gene Name Type of molecule (high vs low risk) ACSL6 ACSL6 acyl-CoA synthetase long-chain family member 6 -1,053 enzyme ACTRT2 ACTRT2 actin-related protein T2 -1,207 other ALKBH3 ALKBH3 alkB, alkylation repair homolog 3 (E. coli) 1,016 enzyme ARMC3 ARMC3 armadillo repeat containing 3 -1,250 other ARPC5 ARPC5 actin related protein 2/3 complex, subunit 5, 16kDa 1,045 other ATG4C ATG4C ATG4 autophagy related 4 homolog C (S. cerevisiae) -1,233 peptidase C14orf70 C14ORF70 chromosome 14 open reading frame 70 1,025 other C17orf78 C17ORF78 chromosome 17 open reading frame 78 -1,418 other C1orf88 C1ORF88 chromosome 1 open reading frame 88 -1,055 other C5orf34 C5ORF34 chromosome 5 open reading frame 34 -1,085 other CASC2 CASC2 cancer susceptibility candidate 2 1,021 other transcription CIAO1 CIAO1 cytosolic iron-sulfur protein assembly 1 homolog (S. cerevisiae) -1,430 regulator COG5 COG5 component of oligomeric golgi complex 5 -1,176 transporter COL17A1 COL17A1 collagen, type XVII, alpha 1 -1,196 other COX4I1 COX4I1 cytochrome c oxidase subunit IV isoform 1 -1,123 enzyme CPSF3 CPSF3 cleavage and polyadenylation specific factor 3, 73kDa -1,380 other CRYBB1 CRYBB1 crystallin, beta B1 -1,214 other DAPK3 DAPK3 death-associated protein kinase 3 -1,063 kinase G-protein coupled DARC DARC Duffy blood group, chemokine receptor 1,024 receptor DIDO1 DIDO1 death inducer-obliterator 1 1,027 other DNAH17 DNAH17 dynein, axonemal, heavy -

An Investigation of the Antigastric Cancer Effect in Tumor Microenvironment of Radix Rhei Et Rhizome: a Network Pharmacology Study
Hindawi Evidence-Based Complementary and Alternative Medicine Volume 2021, Article ID 9913952, 9 pages https://doi.org/10.1155/2021/9913952 Research Article An Investigation of the Antigastric Cancer Effect in Tumor Microenvironment of Radix Rhei Et Rhizome: A Network Pharmacology Study Xinmiao Wang ,1 Guanghui Zhu ,1,2 Haoyu Yang ,1,2 Ruike Gao ,1 Zhe Wu ,1 Ying Zhang,1 Xiaoyu Zhu,1 Xiaoxiao Zhang,1 and Jie Li 1 1Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing 100053, China 2Beijing University of Traditional Chinese Medicine, Beijing 100029, China Correspondence should be addressed to Jie Li; [email protected] Received 27 March 2021; Revised 20 May 2021; Accepted 11 June 2021; Published 24 June 2021 Academic Editor: Yibin Feng Copyright © 2021 Xinmiao Wang et al. +is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background. Tumor microenvironment (TME) takes a vital effect on the occurrence and development of cancer. Radix Rhei Et Rhizome (RRER, Da-Huang in pinyin), a classical Chinese herb, has been widely used in gastric cancer (GC) for many years in China. However, inadequate systematic studies have focused on the anti-GC effect of RRER in TME. +is study intended to uncover the mechanism of it by network pharmacology. Methods. We collected compounds and targets of RRER from traditional Chinese medicine system pharmacology database and analysis platform (TCMSP) and SwissTargetPrediction. GC targets were obtained from GeneCards. Protein- protein interaction (PPI) network and RRER-GC-target network were built by STRING and Cytoscape 3.2.1.