Bridion, INN-Sugammadex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Comparative Study of Different Doses of Rocuronium Bromide For
MedDocs Publishers Annals of Anesthesia and Pain Medicine Open Access | Research Article Comparative study of different doses of rocuronium bromide for endotracheal intubation Nidhi V Sardhara1*; Sonal A Shah2; Dhaval P Pipaliya3; Shivansh Gupta3 1SVP hospital, 13th floor, Room no 13125, Near Ellis bridge, Ahmedabad, Gujarat-380006 2Assistant Professor, Department of anesthesia, Smt. NHL Municipal Medical College, Ahmedabad, Gujarat, India 3First year Resident, Department of anesthesia, Smt. NHL Municipal Medical College, Ahmedabad, Gujarat, India *Corresponding Author(s): Nidhi V Sardhara Abstract SVP hospital, 13th floor, Room no 13125, Near Ellis Background: Endotracheal intubation is one of such bridge, Ahmedabad, Gujarat-380006 development without which general anesthesia cannot be Tel: +91-9428-44-7878, Fax: 7926-57-8452 considered safe for any major surgery particularly head and Email: [email protected] neck, thoracic and abdominal surgeries. Ever since the ad- vent of anesthesia, anesthesiologists have been in search of an ideal muscle relaxants which can provide ideal intu- bating conditions in ultrashort duration with minimal side effects. Rocuronium bromide provides fast onset of action, Received: Mar 11, 2020 an intermediate duration of action and rapid recovery, good Accepted: Apr 24, 2020 to excellent intubating conditions at doses having minimal Published Online: Apr 30, 2020 or no haemodynamic changes. Present study is to compare the effect of different doses (0.6 mg/kg, 0.9 mg/kg, 1.2 mg/ Journal: Annals of Anesthesia and Pain Medicine kg) of Rocuronium bromide for endotracheal intubation at Publisher: MedDocs Publishers LLC 60 seconds. Online edition: http://meddocsonline.org/ Material and methods: This study was carried out by Copyright: © Sardhara NV (2020). -

12-6202 Document: 01018895054 Date Filed: 08/10/2012 Page: 1 FILED United States Court of Appeals UNITED STATES COURT of APPEALS Tenth Circuit
Appellate Case: 12-6202 Document: 01018895054 Date Filed: 08/10/2012 Page: 1 FILED United States Court of Appeals UNITED STATES COURT OF APPEALS Tenth Circuit FOR THE TENTH CIRCUIT August 10, 2012 Elisabeth A. Shumaker Clerk of Court MICHAEL EDWARD HOOPER, Plaintiff-Appellant, v. No. 12-6202 (D.C. No. 5:12-cv-00758-M) JUSTIN JONES, Director DOC; (W.D. Okla.) RANDALL G. WORKMAN, Warden; DOES, Unknown Executioners, Defendants-Appellees. ORDER AND JUDGMENT* Before MURPHY, O’BRIEN, and HOLMES, Circuit Judges. Michael Edward Hooper, an Oklahoma state prisoner scheduled for execution by lethal injection on August 14, 2012, appeals from the district court’s order * After examining the briefs and appellate record, this panel has determined unanimously that oral argument would not materially assist the determination of this appeal. See Fed. R. App. P. 34(a)(2); 10th Cir. R. 34.1(G). The case is therefore ordered submitted without oral argument. This order and judgment is not binding precedent, except under the doctrines of law of the case, res judicata, and collateral estoppel. It may be cited, however, for its persuasive value consistent with Fed. R. App. P. 32.1 and 10th Cir. R. 32.1. Appellate Case: 12-6202 Document: 01018895054 Date Filed: 08/10/2012 Page: 2 denying his motion for a preliminary injunction seeking to stay his execution. Exercising jurisdiction under 28 U.S.C. § 1292(a)(1), we AFFIRM.1 I Mr. Hooper was tried and convicted on three counts of first-degree murder and sentenced to death. See Hooper v. State, 947 P.2d 1090 (Okla. -

Quantitative Analysis of the Aminosteroidal Muscle Relaxant
J. Ecophysiol. Occup. Hlth. 3 & 4 (2013) 29-37 ® 2013 The Academy of Environmental Biology, India Quantitative Analysis of the Aminosteroidal Muscle Relaxant Vecuronium in Rat Liver and Kidneys using Ion-pairing and Liquid Chromatography-Tandem Mass Spectrometry Risha Jasmine Nathan*, P. Sharma, Lily Saroj Nathan1 Forensic Chemistry and Toxicology, Lok Nayak Jayaprakash Narayan National Institute of Criminology, New Delhi 1Ewing Christian College, University of Allahabad Abstract : Muscle relaxants are occasionally encountered in forensic toxicological cases for identification and quantitation. Due to unavailability of human samples, the analysis of the muscle relaxant vecuronium was done using post-mortem rat visceral samples. Rats were injected with bolus overdose of the drug and the post mortem samples were subject to ion-pair extraction with metanil yellow dye. Method for identification of the drug by LC-MS-MS was established first with secondary standard aqueous vecuronium solutions and then applied on the questioned samples. Quantitation was done to find the quantity of the drug in liver and kidney samples. Keywords : Aminosteroid, Muscle relaxant Vecuronium, Liquid Chromatography-Tandem Mass Spectrometry. Introduction ventilation is made available to the patient so as Muscle relaxants, better known as neuro- to maintain respiration as these drugs may also muscular-blocking drugs, are a class of drugs paralyze the diaphragm at the therapeutic dose that block neuromuscular transmission at required (Dripps, 1959). Despite taking neuromuscular junction which causes paralysis precautions, neuromuscular blocking drugs of the affected skeletal muscles. Of the two have been reported to have inadvertently classes, non-depolarizing neuromuscular caused deaths of patients by accident agents work without depolarizing the motor end (Newsletters, Nurse Advise, 2006). -

Product Monograph Vecuronium Bromide For
PRODUCT MONOGRAPH PrVECURONIUM BROMIDE FOR INJECTION Non-depolarizing Skeletal Neuromuscular Blocking Agent Pharmaceutical Partners of Canada Inc. Date of Preparation: 45 Vogell Road, Suite 200 January 15, 2008 Richmond Hill, ON L4B 3P6 Control No.: 119276 PRODUCT MONOGRAPH PrVECURONIUM BROMIDE FOR INJECTION Non-depolarizing Skeletal Neuromuscular Blocking Agent THIS DRUG SHOULD BE ADMINISTERED ONLY BY ADEQUATELY TRAINED INDIVIDUALS FAMILIAR WITH ITS ACTIONS, CHARACTERISTICS AND HAZARDS ACTIONS AND CLINICAL PHARMACOLOGY Vecuronium Bromide for Injection is a non-depolarizing neuromuscular blocking agent of intermediate duration possessing all of the characteristic pharmacological actions of this class of drugs (curariform). It acts by competing for cholinergic receptors at the motor end-plate. The antagonism to acetylcholine is inhibited and neuromuscular block reversed by acetylcholinesterase inhibitors such as neostigmine. Vecuronium Bromide for Injection is about 1/3 more potent than pancuronium; the duration of neuromuscular blockade produced by Vecuronium Bromide for Injection is shorter than that of pancuronium at initially equipotent doses. The time to onset of paralysis decreases and the duration of maximum effect increases with increasing Vecuronium Bromide for Injection doses. The use of a peripheral nerve stimulator is of benefit in assessing the degree of muscular relaxation. The ED90 (dose required to produce 90% suppression of the muscle twitch response with balanced anesthesia) has averaged 0.057 mg/kg (0.049 to 0.062 mg/kg in various studies). An initial Vecuronium Bromide for Injection dose of 0.08 to 0.10 mg/kg generally produces first depression of twitch in approximately 1 minute, good or excellent intubation conditions within 2.5 to 3 minutes, and maximum neuromuscular blockade within 3 to 5 minutes of injection in Vecuronium Bromide for Injection, Product Monograph Page 2 of 28 most patients. -
PMBJP Product.Pdf
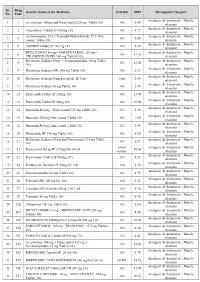
Sr. Drug Generic Name of the Medicine Unit Size MRP Therapeutic Category No. Code Analgesic & Antipyretic / Muscle 1 1 Aceclofenac 100mg and Paracetamol 325 mg Tablet 10's 10's 8.00 relaxants Analgesic & Antipyretic / Muscle 2 2 Aceclofenac Tablets IP 100mg 10's 10's 4.37 relaxants Acetaminophen 325 + Tramadol Hydrochloride 37.5 film Analgesic & Antipyretic / Muscle 3 4 10's 8.00 coated Tablet 10's relaxants Analgesic & Antipyretic / Muscle 4 5 ASPIRIN Tablets IP 150 mg 14's 14's 2.70 relaxants DICLOFENAC 50 mg+ PARACETAMOL 325 mg+ Analgesic & Antipyretic / Muscle 5 6 10's 11.30 CHLORZOXAZONE 500 mg Tablets 10's relaxants Diclofenac Sodium 50mg + Serratiopeptidase 10mg Tablet Analgesic & Antipyretic / Muscle 6 8 10's 12.00 10's relaxants Analgesic & Antipyretic / Muscle 7 9 Diclofenac Sodium (SR) 100 mg Tablet 10's 10's 6.12 relaxants Analgesic & Antipyretic / Muscle 8 10 Diclofenac Sodium 25mg per ml Inj. IP 3 ml 3 ml 2.00 relaxants Analgesic & Antipyretic / Muscle 9 11 Diclofenac Sodium 50 mg Tablet 10's 10's 2.90 relaxants Analgesic & Antipyretic / Muscle 10 12 Etoricoxilb Tablets IP 120mg 10's 10's 33.00 relaxants Analgesic & Antipyretic / Muscle 11 13 Etoricoxilb Tablets IP 90mg 10's 10's 25.00 relaxants Analgesic & Antipyretic / Muscle 12 14 Ibuprofen 400 mg + Paracetamol 325 mg Tablet 10's 15's 5.50 relaxants Analgesic & Antipyretic / Muscle 13 15 Ibuprofen 200 mg film coated Tablet 10's 10's 1.80 relaxants Analgesic & Antipyretic / Muscle 14 16 Ibuprofen 400 mg film coated Tablet 10's 15's 3.50 relaxants Analgesic & Antipyretic -

Vecuronium Bromide As Control
A COMPARATIVE STUDY TO FIND AN IDEAL INTUBATING DOSE OF INJ. ROCURONIUM BROMIDE USING INJ. VECURONIUM BROMIDE AS CONTROL. Dissertation submitted In partial fulfillment for the award of M.D DEGREE EXAMINATION M.D. ANAESTHESIOLOGY & CRITICAL CARE BRANCH X GOVT KILPAUK MEDICAL COLLEGE AND HOSPITAL SUBMITTED TO THE TAMILNADU DR. MGR MEDICAL UNIVERSITY CHENNAI APRIL – 2011 1 DECLARATION I, Dr. D.Shunmuga Priya, solemnly declare that the dissertation, “A COMPARATIVE STUDY TO FIND AN IDEAL INTUBATING DOSE OF INJ. ROCURONIUM BROMIDE USING INJ. VECURONIUM BROMIDE AS CONTROL” is a bonafide work done by me in the Department of Anaesthesiology and Critical Care, Government Kilpauk, Medical College, Chennai under the able guidance of Prof. Dr. P.S. Shanmugam, MD., DA., Professor and HOD, Department of Anaesthesiology and Critical Care, Government Kilpauk Medical College, Chennai. Place : Chennai Date : (Dr. D.SHUNMUGA PRIYA) 5 CERTIFICATE This is to certify that this dissertation titled “A COMPARATIVE STUDY TO FIND AN IDEAL INTUBATING DOSE OF INJ. ROCURONIUM BROMIDE USING INJ. VECURONIUM BROMIDE AS CONTROL” has been prepared by Dr. D.SHUNMUGA PRIYA under my supervision in the Department of Anaesthesiology and Critical Care, Government Kilpauk Medical College, Chennai during the academic period 2008-2011 and is being submitted to the Tamil Nadu Dr. M.G.R. Medical University, Chennai in partial fulfillment of the University regulation for the award of the Degree of Doctor of Medicine (MD Anaesthesiology and Critical Care) and her dissertation is a bonafide work. Prof..Dr. V.Kanagasabai, M.D Prof. Dr. P.S.Shanmugam, M.D., D.A. DEAN Professor & HOD, Government Kilpauk Medical College Department of Anaesthesiology and & Hospital, Critical Care, Chennai. -

Vecureinj.Pdf
NEW ZEALAND DATA SHEET VECURE 1. Product Name VECURE 10 mg, powder for injection. 2. Qualitative and Quantitative Composition VECURE 10 mg, 1 vial contains 10 mg vecuronium bromide. When reconstituted with 5 mL water for injection corresponds to 2 mg vecuronium bromide per mL in a clear almost clear isotonic solution. For the full list of excipients, see section 6.1 3. Pharmaceutical Form White, freeze dried powder for injection. Following reconstitution with solvent (water for injection) the resultant solution is isotonic and has a pH of 4. 4. Clinical Particulars 4.1 Therapeutic indications Vecure is indicated as an adjunct to general anaesthesia to facilitate tracheal intubation and to provide skeletal muscle relaxation during surgery. 4.2 Dose and method of administration Dosage Like other neuromuscular blocking agents, VECURE should only be administered by, or under supervision of, experienced clinicians who are familiar with the action and use of these agents. Like with other neuromuscular blocking agents, the dosage of VECURE should be individualised in each patient. The anaesthetic method used, the expected duration of surgery, the possible interaction with other medicines that are administered before or during anaesthesia and the condition of the patient should be taken into account when determining the dose. The use of an appropriate neuromuscular monitoring technique is recommended to monitor neuromuscular block and recovery. Inhalational anaesthetics do potentiate the neuromuscular blocking effects of VECURE. This potentiation however, becomes clinically relevant in the course of anaesthesia, when the volatile agents have reached the tissue concentrations required for this interaction. Consequently, adjustments with VECURE should be made by administering smaller maintenance doses at less frequent intervals or by using lower infusion rates of VECURE during long lasting procedures (longer than 1 hour) under inhalational anaesthesia (see section 4.5). -

Geisinger Lewistown Hospital Published: March 25, 2019
Geisinger Lewistown Hospital Published: March 25, 2019 DESCRIPTION CHARGE Fine needle aspiration; without imaging guidance $ 607.00 Fine needle aspiration; without imaging guidance $ 286.00 Fine needle aspiration; with imaging guidance $ 2,218.00 Fine needle aspiration; with imaging guidance $ 1,691.00 Placement of soft tissue localization device(s) (eg, clip, metallic pellet, wire/needle, radioactive seeds), percutaneous, including imaging guidance; first lesion $ 1,979.00 Placement of soft tissue localization device(s) (eg, clip, metallic pellet, wire/needle, radioactive seeds), percutaneous, including imaging guidance; each $ 1,385.00 additional lesion (List separately in addition to code for primary procedure) Incision and drainage of abscess (eg, carbuncle, suppurative hidradenitis, cutaneous or subcutaneous abscess, cyst, furuncle, or paronychia); simple or single $ 657.00 Incision and drainage of abscess (eg, carbuncle, suppurative hidradenitis, cutaneous or subcutaneous abscess, cyst, furuncle, or paronychia); complicated or $ 986.00 multiple Incision and drainage of pilonidal cyst; simple $ 657.00 Incision and drainage of pilonidal cyst; complicated $ 3,726.00 Incision and removal of foreign body, subcutaneous tissues; simple $ 1,694.00 Incision and removal of foreign body, subcutaneous tissues; complicated $ 4,710.00 Incision and drainage of hematoma, seroma or fluid collection $ 3,470.00 Puncture aspiration of abscess, hematoma, bulla, or cyst $ 1,272.00 Puncture aspiration of abscess, hematoma, bulla, or cyst $ 657.00 Incision -

Rocuronium Bromide PRODUCT DATA SHEET Issue Date 01/06/2020
Rocuronium Bromide PRODUCT DATA SHEET issue date 01/06/2020 Product Name: Rocuronium Bromide Product Number: R045 CAS Number: 119302-91-9 Molecular Formula: C32H53BrN2O4 Molecular Weight: 609.68 Form: Powder Appearance: Off-white to yellowish powder Solubility: Soluble in water, ethanol, and DMSO. Source: Synthetic Storage Conditions: ≤30°C Description: Rocuronium Bromide is the bromide salt form of Rocuronium, a fumarate, aminosteroid type non-depolarizing neuromuscular blocking agent that relaxes the skeletal muscle. It was introduced in 1994, and has a similar pharmacokinetic profile to Vecuronium, as it is a derivative of the 3-hydroxy metabolite of Vecuronium. It competes with acetylcholine in binding to cholinergic receptors at neuromuscular junctions. Rocuronium Bromide is soluble in water, ethanol, and DMSO. Mechanism of Action: Rocuronium Bromide competes with acetylcholine and binds to cholinergic receptors at neuromuscular junctions. It binds to nicotinic receptors in the neuromuscular junction. It is classified as a neuromuscular nondepolarizing agent since it does not cause depolarization of the motor end plate. References: Anzenbacherova et al (2015) Interaction of Rocuronium with human liver cytochromes P450. J. Pharmacol. Sci. 127(2):190-195 PMID 25727956 Cembala TM, Sherwin JD, Tidmarsh MD, Appadu BL and Lambert DG (1998) Interaction of neuromuscular blocking drugs with recombinant human m1-m5 muscarinic receptors expressed in Chinese hamster ovary cells. B. J. Pharmacol. 125(5):1088-1094 PMID 9846649 Koksal PM and Gürbüzel M(2015) Analysis of genotoxic activity of ketamine and Rocuronium Bromide using the somatic mutation and recombination test in Drosophila melanogaster. Environ Toxicol Pharmacol. 39(2):628-634 PMID 25682000 Sauer et al (2017) Rocuronium is more hepatotoxic than succinylcholine in vitro. -

COMPARISON of ROCURONIUM BROMIDE and SUCCINYLCHOLINE CHLORIDE for USE DURING RAPID SEQUENCE INTUBATION in ADULTS Ch
DOI: 10.18410/jebmh/2015/672 ORIGINAL ARTICLE COMPARISON OF ROCURONIUM BROMIDE AND SUCCINYLCHOLINE CHLORIDE FOR USE DURING RAPID SEQUENCE INTUBATION IN ADULTS Ch. Penchalaiah1, N. Jagadeesh Babu2, T. Kumar3 HOW TO CITE THIS ARTICLE: Ch. Penchalaiah, N. Jagadeesh Babu, T. Kumar. ”Comparison of Rocuronium Bromide and Succinylcholine Chloride for Use during Rapid Sequence Intubation in Adults”. Journal of Evidence based Medicine and Healthcare; Volume 2, Issue 32, August 10, 2015; Page: 4796-4806, DOI: 10.18410/jebmh/2015/672 ABSTRACT: BACKGROUND AND OBJECTIVE: The goal of rapid sequence intubation is to secure the patients airway smoothly and quickly, minimizing the chances of regurgitation and aspiration of gastric contents. Traditionally succinylcholine chloride has been the neuromuscular blocking drug of choice for use in rapid sequence intubation because of its rapid onset of action and profound relaxation. Succinylcholine chloride remains unsurpassed in providing ideal intubating conditions. However the use of succinylcholine chloride is associated with many side effects like muscle pain, bradycardia, hyperkalaemia and rise in intragastric and intraocular pressure. Rocuronium bromide is the only drug currently available which has the rapidity of onset of action like succinylcholine chloride. Hence the present study was undertaken to compare rocuronium bromide with succinylcholine chloride for use during rapid sequence intubation in adult patients. METHODOLOGY: The study population consisted of 90 patients aged between 18-60 years posted for various elective surgeries requiring general anaesthesia. Study population was randomly divided into 3 groups with 30 patients in each sub group. 1. Group I: Intubated with 1 mg kg-1 of succinylcholine chloride (n=30). -

INTUBATING CONDITIONS and INJECTION PAIN - Cisatracurium Or Rocuronium Versus Rocuronium-Cisatracurium Combination
INTUBATING CONDITIONS AND INJECTION PAIN - Cisatracurium or Rocuronium versus Rocuronium-Cisatracurium Combination - * * AHED ZEIDAN ,NAZIH NAHLE , ** *** HILAL MAALIKI AND ANIS BARAKA Summary The present report evaluates the incidence of pain on intravenous injection and the condition of tracheal intubation at one minute following the administration of cisatracurium or rocuronium versus rocuronium- cisatracurium combination. We studied 60 patients, ASA 1, aged 18-60 years, undergoing elective surgical procedures. The patients were randomly assigned to 3 groups who received intravenously either 0.15 mg/kg cisatracurium [2ED95], 0,6 mg rocuronium [2ED95] or a combination of 0.075 mg/kg cisatracurium [1ED95], plus 0.3 mg rocuronium [1ED95]. In the awake patients, the pain on injection of muscle relaxant was assessed on a four point scale (none, mild, moderate, severe). Administration of the relaxant was followed by 1-2 mg/kg of lidocaine and 2 mg/kg propofol. Oro- tracheal intubation was performed 60 seconds following the administration of the relaxant. The intubating conditions were assessed and rated as excellent, good, fair or poor. * MD, Staff Anaesthesiologist, Department of Anaesthesiology, Sahel General Hospital, Beirut, Lebanon. **MD Staff Anaesthesiologist, Department of Anaesthesiology, Beirut Governmental University Hospital, Beirut, Lebanon. *** MD, FRCA, Professor and and Chairman, Department of Anesthesiology, American University of Beirut Medical Center, Beirut, Lebanon. Corresponding Author: Ahed Zeidan, Department of Anaesthesiology, Sahel General Hospital, Airport Ave. P.O. Box: 99/25-Ghobeiry, Beirut-Lebanon. Phone: 961-1-858333. Fax: 961-1-840146, E-mail: [email protected]. 879 M.E.J. ANESTH 18 (5), 2006 880 AHED ZEIDAN ET. AL The administration of 2ED95 cisatracurium resulted in poor intubating conditions at 60s, without pain on injection. -

Anesthetic Considerations for the Parturient with Idiopathic Intracranial Hypertension Levi G
University of North Dakota UND Scholarly Commons Nursing Capstones Department of Nursing 7-12-2018 Anesthetic Considerations for the Parturient with Idiopathic Intracranial Hypertension Levi G. La Porte Follow this and additional works at: https://commons.und.edu/nurs-capstones Recommended Citation La Porte, Levi G., "Anesthetic Considerations for the Parturient with Idiopathic Intracranial Hypertension" (2018). Nursing Capstones. 187. https://commons.und.edu/nurs-capstones/187 This Independent Study is brought to you for free and open access by the Department of Nursing at UND Scholarly Commons. It has been accepted for inclusion in Nursing Capstones by an authorized administrator of UND Scholarly Commons. For more information, please contact [email protected]. Running head: SAFETY OF SUGAMMADEX AND NEOSTIGMINE 1 The Safety and Efficacy in Reversal of Neuromuscular Blockades with Sugammadex versus Neostigmine by Nicole L. Landen Bachelor of Science in Nursing, University of Minnesota, 2014 An Independent Study Submitted to the Graduate Faculty of the University of North Dakota in partial fulfillment of the requirements for the degree of Master of Science Grand Forks, North Dakota December 2019 SAFETY OF SUGAMMADEX AND NEOSTIGMINE 2 PERMISSION Title The Safety and Efficacy in Reversal of Neuromuscular Blockades with Sugammadex versus Neostigmine Department Nursing Degree Master of Science In presenting this independent study in partial fulfillment of the requirements for a graduate degree from the University of North Dakota, I agree that the College of Nursing and Professional Disciplines of this University shall make it freely available for inspection. I further agree that permission for extensive copying or electronic access for scholarly purposes may be granted by the professor who supervised my independent study work or, in her absence, by the chairperson of the department or the dean of the School of Graduate Studies.