Immunochemical Determination of Caffeine and Carbamazepine in Complex Matrices Using Fluorescence Polarization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PDF Product Guide
05/24/21 To Place an Order, Call (207)947-0321 Fax: (207)947-0323 ITEM # DESCRIPTION PACK ITEM # DESCRIPTION PACK COCKTAIL MIXES COFFEE FLAVORED 14678 ROLAND OLIVE JUICE DIRTY*MARTIN 12/25.4OZ 10927 MAINE'S BEST COFFEE JAMAICAN ME CRAZY 2 24/2.25OZ 2008 OCEAN SPRAY DRINK MIX BLOODY MARY 12/32 OZ 11282 MAINE'S BEST COFFEE VACATIONLND VANILLA 24/2.25OZ 26628 MAYSON'S MARGARITA MIX ON THE ROCKS 4/1 GAL 11516 MAINE'S BEST COFFEE HARBORSIDE HAZELNUT 24/2.25OZ 26633 MAYSON'S MARGARITA MIX FOR FROZEN 4/1 GAL 83931 NewEngland COFFEE REG FRNCH VAN CRAZE 24/2.5OZ 26634 MAYSON'S MARGARITA STRAWBERRY PUREE 4/1 GAL 83933 NewEngland COFFEE REG HAZELNUT CRAZE 24/2.5OZ 26637 MAYSON'S MARGARITA RASPBERRY PUREE 4/1 GAL 83939 NewEngland COFFEE REG PUMPKIN SPICE 24/2.5OZ 26639 MAYSON'S MARGARITA PEACH PUREE 4/1 GAL 83940 NewEngland COFFEE REG CINNAMON STICKY B 24/2.5OZ 26640 MAYSON'S MARGARITA WATERMELON PUREE 4/1 GAL 26724 MAYSON'S MARGARITA MANGO PUREE 4/1 GAL 83831 Packer DRINK MIX LEMON POWDER 12/1GAL COFFEE REGULAR 83890 COCO LOPEZ DRINK MIX CREAM COCONUT 24/15oz 10444 MAXWELL HOUSE COFFEE HOTEL & REST 112/1.6OZ 84011 Rose's SYRUP GRENADINE 12/1LTR 10479 Folgers COFFEE LIQ 100% COLOMBIAN 2/1.25L 10923 MAINE'S BEST COFFEE REG ACADIA BLEND 2O 42/2OZ 10924 MAINE'S BEST COFFEE REG COUNTY BLEND 1. 42/1.5OZ CAPPUCCINO 10930 MAINE'S BEST COFFEE REG DWNEAST DARK 2. 24/2.25OZ 90343 Int Coffee CAPPUCCINO FRENCH VAN 6/2LB 1410 Maxwellhse COFFEE REGULAR MASTERBLEND 64/3.75 OZ 23529 MAINE'S BEST COFFEE SEBAGO BLEND 42/2.25OZ 23531 NEW ENGLAND COFFEE EXTREME KAFFEINE -

Ice Cream Catalog
800-642-6124 978-948-7276 • Fax 978-948-6181 www.jstonediamondfoods.com Custom Packaging • Ice Cream & Confectionery Ingredients FOSS Toppings and Syrups FS-10-001PCH New England Fudge 6/.5 GL PCHS FS-10-001PL New England Fudge 46LBS FS-10-002PCH Milk Fudge 6/.5 GL PCHS FS-10-010 Wicked Good Fudge 6/#10 Can FS-10-010PCH Wicked Good Fudge 6/.5GL PCHS FS-11-006GL Foss Chocolate Syrup 4/GL FS-11-004 Lighthouse Chocolate Syrup 4/GL FS-13-007 Caramel Topping 6/#10 Can FS-13-008 Butterscotch Topping 6/#10 Cans FS-13-009PCH Butterscotch Topping 12/32 OZ PCHS FS-14-028PCH Pineapple Topping 12/32 OZ PCHS FS-14-018PCH Blueberry Topping 12/32 OZ PCHS FS-18-033 RTU Vanilla Syrup 4/GL FS-18-034 RTU Coffee Syrup 4/GL FS-18-035 RTU Strawberry Syrup 4/GL FS-18-037 RTU Cherry Syrup 4/GL FS-18-039 RTU Raspberry Syrup 4/GL FS-18-041 RTU Lime Rickey Syrup 4/GL Retail FS-10-021RET Wicked Good Fudge 6/ 9 OZ Jars FS-13-000RET Caramel 6/ 9 OZ Jars Ingredients FS-20-301 Foss Midnight Chocolate ICP 6/#10 Cans (HH-167) FS-20-303 #355 Real Rich Chocolate ICP 6/#10 Cans FS-20-304 Extra Strength Chocolate ICP 6/#10 Cans FS-20-304P Extra Strength Chocolate Pail 50LBS FS-20-315 Butter Pecan Base 6/#10 Cans FS-20-318 Lite Orange-Pineapple 6/#10 Cans FS-20-310PCH Egg Custard Base 12/Qt PCHS 2 FOSS ingredients continued FS-20-320 Black Raspberry 6/#10 Cans FS-20-322 Fudge for Ice Cream 6/#10 Cans FS-20-332 NY Style Cheesecake 6/#10 Cans FS-20-346 Maine Wild Blueberry ICB 6/#10 Cans FS-20-361 Natural Black Rasp Flavor-4/1 GL FS-21-412P Wicked Good Fudge Variegate 46LBS -
Kosher Nosh Guide Summer 2020
k Kosher Nosh Guide Summer 2020 For the latest information check www.isitkosher.uk CONTENTS 5 USING THE PRODUCT LISTINGS 5 EXPLANATION OF KASHRUT SYMBOLS 5 PROBLEMATIC E NUMBERS 6 BISCUITS 6 BREAD 7 CHOCOLATE & SWEET SPREADS 7 CONFECTIONERY 18 CRACKERS, RICE & CORN CAKES 18 CRISPS & SNACKS 20 DESSERTS 21 ENERGY & PROTEIN SNACKS 22 ENERGY DRINKS 23 FRUIT SNACKS 24 HOT CHOCOLATE & MALTED DRINKS 24 ICE CREAM CONES & WAFERS 25 ICE CREAMS, LOLLIES & SORBET 29 MILK SHAKES & MIXES 30 NUTS & SEEDS 31 PEANUT BUTTER & MARMITE 31 POPCORN 31 SNACK BARS 34 SOFT DRINKS 42 SUGAR FREE CONFECTIONERY 43 SYRUPS & TOPPINGS 43 YOGHURT DRINKS 44 YOGHURTS & DAIRY DESSERTS The information in this guide is only applicable to products made for the UK market. All details are correct at the time of going to press but are subject to change. For the latest information check www.isitkosher.uk. Sign up for email alerts and updates on www.kosher.org.uk or join Facebook KLBD Kosher Direct. No assumptions should be made about the kosher status of products not listed, even if others in the range are approved or certified. It is preferable, whenever possible, to buy products made under Rabbinical supervision. WARNING: The designation ‘Parev’ does not guarantee that a product is suitable for those with dairy or lactose intolerance. WARNING: The ‘Nut Free’ symbol is displayed next to a product based on information from manufacturers. The KLBD takes no responsibility for this designation. You are advised to check the allergen information on each product. k GUESS WHAT'S IN YOUR FOOD k USING THE PRODUCT LISTINGS Hi Noshers! PRODUCTS WHICH ARE KLBD CERTIFIED Even in these difficult times, and perhaps now more than ever, Like many kashrut authorities around the world, the KLBD uses the American we need our Nosh! kosher logo system. -

Packaging&Design
FAB SEVENTH INTERNATIONAL FOOD AND BEVERAGE Creative Excellence Awards WINNERS 2005 Packaging&Design The Retailers Boxer, Birmingham McDonalds i'm lovin' it Bags McDonalds Nomination Red Design Consultants, Athens Bakaliko Range Bakaliko Nomination Pet Foods No Nominations Confectionary&Snacks Turner Duckworth, London Dried Vine Fruit Range Waitrose Nomination Ziggurat Brand Consultants, London Jonathan Crisps Range Natural Crisps Ltd FAB Williams Murray Hamm, London Little Notions Range Nestlé Nomination Ruiz & Company, Barcelona La Fondue Range Chocolat Factory Nomination Health Foods No Nominations Dairy Products Red Design Consultants, Athens Imagine Ice Cream Range Delta Ice Cream Nomination Williams Murray Hamm, London Hill Station Ice Cream Range Hill Station Nomination Reach, Bristol Rocombe Farm Fresh Ice Cream Range Rocombe Farm Fresh Ice Cream FAB Photography No Nominations Baking&Sweet Foods Turner Duckworth, London Mr Kipling Range Manor Bakeries Nomination Savoury Foods Design Bridge, London Toro Toro Ethnic Range Rieber & Son Ziggurat Brand Consultants, London Mrs Massey Range Mrs Massey FAB Pearlfisher, London Ready Meals Range Waitrose Nomination Alcoholic Drinks Hornall Anderson Design Works, Seattle W'05 IPA W'05 IPA Widmer Brothers Nomination Design Bridge, London The Beer to Dine for The Beer to Dine for Greene King FAB Design Bridge, London Sabai Cooler Sabai Siam Wineries Nomination Williams Murray Hamm, London Fallen Vodka Batch No 1, 2, 3 Glenmorangie Nomination Non-Alcoholic Drinks Williams Murray Hamm, London -

AUGUST ICE CREAM PROMOTIONS R&R TAKEHOME 82205 Nestle Milkybar Oval - P.M
Consort Way Woodford Trading Estate Burgess Hill Southend Road West Sussex Woodford RH15 9TJ Woodford Green Greater London IG8 8HF Tel: 01444 870 111 Tel: 020 8550 4486 www.consortfrozenfoods.co.uk www.directwholesalefoods.com FWG PRODUCT CASE LONDIS LONDIS RSP LONDIS MARGIN CODE NAME SIZE PRICE INC VAT EXC VAT AUGUST ICE CREAM PROMOTIONS R&R TAKEHOME 82205 Nestle Milkybar Oval - P.M. £2.50 8 £11.92 £2.25 20.53% 76970 Nestle Smarties Oval - P.M. £2.50 8 £11.92 £2.25 20.53% 77728 Nestle Rolo Oval 800ml - P.M. £2.50 8 £11.92 £2.25 20.53% 78189 Fab Strawberry - P.M. £2.00 8 £9.54 £1.75 18.32% WALL’S BUY ALL 5 - GET CALIPPO ORANGE WORTH £21.60 AT RSP FREE 86815 Cornetto Strawberry 24 £14.92 £1.20 37.83% 73807 Magnum White 20 £16.64 £1.60 37.60% 88732 Magnum Infinity 20 £18.20 £1.75 37.60% 73556 Magnum Classic 20 £16.64 £1.60 37.60% 83006 Twister 35 £16.23 £0.90 38.16% FREE! 76270 Calippo Orange FOC 24 £- £0.90 100.00% WALL’S BUY ALL 5 - GET SOLERO EXOTIC WORTH £30.00 AT RSP FREE 81821 Magnmum Double Caramel 20 £16.64 £1.60 37.60% 74546 Magnum Almond 20 £16.64 £1.60 37.60% 86582 Haribo Push-Up 30 £20.93 £1.35 37.99% 76324 Feast 35 £17.94 £1.00 38.48% FREE! 88122 Calippo Shots 24 £13.64 £1.10 38.00% 83863 Solero Exotic FOC 25 £- £1.20 100.00% R&R BUY BOTH - GET FAB STRAWBERRY WORTH £28.80 AT RSP FREE 87610 Flake Cone 24 £20.48 £1.60 36.00% 84113 Cadbury's Dairy Milk Swirl 24 £20.48 £1.60 36.00% 76773 Fab Strawberry FOC 32 £- £0.90 100.00% FREE! R&R BUY ANY 7 - GET FRUIT SHOOT ORANGE AND SMARTIES POP UP WORTH £50.00 AT RSP FREE 82559 Cadbury's -
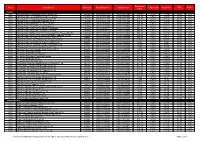
Code Description Unit Size Retail Barcode Unit Barcode Trade List
Trade List Code Description Unit Size Retail Barcode Unit Barcode % Discount Your Price RSP POR% Price PIZZAS 4665 Chicago Town 2 Deep Dish Chicken Club £2.00 12x320g 5019312081442 5019312441802 £19.23 6.40% £18.00 £2.00 25% 5141 Chicago Town 2 Deep Dish Four Cheese £2.00 12x310g 5019312081343 5019312431803 £19.23 6.40% £18.00 £2.00 25% 4616 Chicago Town 2 Deep Dish Mega Meaty £2.00 12x320g 5019312081497 5019312941807 £19.23 6.40% £18.00 £2.00 25% 5139 Chicago Town 2 Deep Dish Pepperoni £2.00 12x320g 5019312081336 5019312331806 £19.23 6.40% £18.00 £2.00 25% 1636 Chicago Town Pizza Kitchen Cheese Medley 10x350g 5019312093803 5019312083903 £24.90 6.40% £23.31 £3.00 22% 1637 Chicago Town Pizza Kitchen Deli Pepperoni 10x355g 5019312093766 5019312673906 £24.90 6.40% £23.31 £3.00 22% 1639 Chicago Town Pizza Kitchen Garden Vegetables 10x350g 5019312093797 5019312973907 £24.90 6.40% £23.31 £3.00 22% 1638 Chicago Town Pizza Kitchen Roasted Chicken 10x385g 5019312093773 5019312773903 £24.90 6.40% £23.31 £3.00 22% 6708 Chicago Town Stuffed Crust 25cm Takeaway Four Cheese Melt £3 10x480g 5019312103809 5019312083019 £24.99 6.40% £23.39 £3.00 22% 6707 Chicago Town Stuffed Crust 25cm Takeaway Pepperoni Plus £3 10x490g 5019312003802 5019312083002 £24.99 6.40% £23.39 £3.00 22% 8556 Chicago Town Takeaway 30cm Classic Crust Cheese 8x490g 5019312082999 5019312992809 £19.99 6.40% £18.71 £3.00 22% 5772 Chicago Town The Deep Pan Double Pepperoni £2.00 6x415g 5019312089271 5019312729801 £9.90 6.40% £9.27 £2.00 23% 5771 Chicago Town The Deep Pan Triple Cheese £2.00 6x405g 5019312089288 5019312829808 £9.90 6.40% £9.27 £2.00 23% 6706 Chicago Town The Thin One Double Pepperoni £2.00 6x305g 5019312089332 5019312339802 £9.99 6.40% £9.35 £2.00 22% 6612 Chicago Town The Thin One Meat Feast £2.00 6x325g 5019312089356 5019312539806 £9.99 6.40% £9.35 £2.00 22% 6705 Chicago Town The Thin One Triple Cheese £2.00 6x305g 5019312089325 5019312239805 £9.99 6.40% £9.35 £2.00 22% 8559 Dr. -

Essentials & Perfect Presents
2 2017 HOMEEssentials & Perfect Presents Shop with confidence! Every purchase comes with our 100% Customer Satisfaction Guarantee. P02-13 Gourmet Delights P14-21 Home Essentials P22-33 Unique Boutique P34-47 Holiday Shoppe P48 Woodwick Candles SHOW COPY Shop & share with confidence! Jalepeno Garlic Taste of Tuscany Gusto Garlic Pesto Cherrydale has been helping groups “just” like ours raise money, since 1907. Over 500 gift items in this catalog and online! Many priced below $10! Cherrydale Fundraising exclusives too! 2429 l Slate 3 Cheeseball ll a ... ba o A nto rc i n Flavors in d a a n h t e u l C r 1 Box B #hosthomepartyfundraiser a lly fo 3235 o Gourmet Delights d -s a Ro l b f al e l s in ur T face H I Clever Cuisine S 3277 SLATE “Home Essentials & Perfect Presents” COASTERS, SET OF 4 Servidor de 2 Niveles AT H Natural slate coasters keep T TABLE OF CONTENTS surfaces safe. Set of 4, 3” x 3”. Non slip feet o keep coasters in place and protect surfaces. P02-13 “Gourmet Delights” Mix, season, prep, serve & eat. 3235 TWO TIERED SLATE SERVER Servidor de 2 Niveles Beautifully handcrafted from natural slate with P14-21 “Home Essentials” Kitchen tools, housewares, etc. stainless steel center handle and non-slip feet. Use chalk to label your selections! 9-7/8” Dia x o TH See P24 to 7-7/8”Dia x 9”H. IS P22-33 “Unique Boutique” Unique, useful , personalize ! subscriptions , items! 2429 CHEESE BALL TRIO exclusives, treats & Mezclas de queso, 3 sabores A great tasting blend of spices come together to form an explosion of flavor when combined P34-47 “Holiday” Shoppe” Heavy roll wrap, decor & more. -

Impulse Ice Cream List Unit Unit Portion/Unit Size Price Cost
Impulse Ice Cream List Unit Unit Portion/Unit Size Price Cost 100% Real Fruit Frenzy 7952 100% REAL Fruit Frenzy Apple & Raspberry 20x105ml 14.33 ^ 0.72 ^ 9638 100% Real Fruit Frenzy Blackcurrant 20x105ml 14.33 ^ 0.72 ^ 4469 100% Real Fruit Frenzy Orange 20x105ml 14.33 ^ 0.72 ^ 100% Real Fruit Yummy Lollies Kids « 7016 Fruitic Apple & Raspberry 50x30ml 14.18 ^ 0.28 ^ « 5412 Yummy 100% Lemon 25x60ml 14.58 ^ 0.58 ^ « 6201 Yummy Apple & Mango 25x60ml 14.58 ^ 0.58 ^ « 5349 Yummy Blackcurrant 25x60ml 14.58 ^ J 0.58 ^ « 1001 Yummy British Apple & Raspberry 25x60ml 14.58 ^ J 0.58 ^ « 4424 Yummy Orange Ice Lolly 25x60ml 14.58 ^ 0.58 ^ « 7217 Yummy Strawberry & Banana 25x60ml 14.58 ^ 0.58 ^ Walls Impulse - Magnum 5105 Magnum Champagne 20x100ml 22.75 ^ 1.14 ^ 1118 Magnum Classic 1x20 20.80 ^ 1.04 ^ 1119 Magnum White 1x20 20.80 ^ J 1.04 ^ 8031 Magnum Almond 1x20 20.80 ^ J 1.04 ^ 9556 Magnum Mint 1x20 20.80 ^ J 1.04 ^ 1328 Magnum Double Caramel 1x20 20.80 ^ 1.04 ^ 7418 Magnum Infinity 1x20 22.75 ^ J 1.14 ^ Wall's Impulse Ice Cream 1323 Cornetto Strawberry 1x24 18.65 ^ 0.78 ^ 1324 Cornetto Classico 1x24 18.65 ^ 0.78 ^ 1325 Cornetto Mint Choc Chip 1x24 18.65 ^ 0.78 ^ 2854 Cornetto Raspberry Cheesecake 24x125ml 23.40 ^ 0.98 ^ 4472 Cornetto Vanilla Shake * 1x24 23.40 ^ 0.98 ^ 3973 PM 1.00 Cornetto Classico 30x90ml 19.50 ^ 0.65 ^ 5549 PM 1.00 Cornetto Strawberry 30x90ml 19.50 ^ 0.65 ^ 7946 Cornetto Choc 'n' Ball 20x160ml 22.75 ^ 1.14 ^ 1333 Feast Original 1x35 22.43 ^ 0.64 ^ 2364 Feast Mint 1x35 22.43 ^ 0.64 ^ 1043 Twister 1x35 20.29 ^ 0.58 ^ 1156 Calippo -

FAB Fabulous Grandfabulous
The FAB Awards is an International Creative and Effectiveness Awards programme focussed entirely on work done for Food And Beverage brands. It recognises the critical contribution that outstanding creative work makes to building brands, it identifies and rewards the leading practitioners from over 60 countries, and it acknowledges their contribution to their Clients and their Agencies. FAB FABulous For the best entry in each category Overall winner of the medium GrandFABulous For the single outstanding entry irrespective of category Now in its 17th year, The FAB Awards has taken entries from many Clients, every leading Agency Network and many important Challenger Agencies from over 60 countries around the world. The FAB Agency of the Year 2014 was AMV BBDO London, Network of the Year was Leo Burnett and other FAB Awards went to 16 different countries, all presented at our Awards Dinner held in the glorious surroundings of The Hurlingham Club in London. The FAB Awards Brand of the Year 2014 was Kit Kat. There is still no other international Awards programme which shares our focus, and The FAB Awards continues to offer Agencies and their Clients a unique opportunity. THE RETAILERS restaurants (both fast food and formal) / hotels / bars and clubs / grocery outlets / off licences / convenience stores / petrol stations / airports & railway outlets HEALTH FOODS low calorie foods / low fat foods / organic foods / slimming products / sweeteners / energy drinks / vitamin and mineral supplements / lowered cholesterol products DAIRY PRODUCTS eggs -

2017 CBF Trifold Numbered Pages (Compressed)
Always Natural: ° No Artificial Colors ° No Hydrogenated Oils ° ° RBGH Free Cream Cheese ° Non-GMO Real Cane Sugar ° ° Non-GMO Flour ° ••• Always Handmade: ° Made in Small Batches ° Great Attention to Consistency ° ° Highest Quality ° ° Co-Branding & Private Label ° Kosher ° SQF Level 2 Certified ° p|360.380.1908 • f|360.384.3673 • [email protected] • 5501 Hovander Road #1 Ferndale | WA 98248 www.chuckanutbay.com Retail Cheesecakes 3” Single Serve Cheesecake This is our best-selling item, a perfect portion for one. With flavors that cover all seasons of the year. This cheesecake is ideal for “grab-n-go” sales. They are handmade in small batches, and crafted as a special dessert for one. Available in 3.5 oz & 4 oz 6 or 12 to a case pack 5” Retail Cheesecake It is an excellent size for smaller families or small gatherings. Offered in unique flavors, like Chocolate Stout as well as our best selling flavors like New York and Strawberry. This 16 ounce cake serves four with perfect 4 oz servings. Available in 16 oz 6 to a case pack 7” Retail Cheesecake A great choice for larger families or group gatherings. It is perfect to take home for any event. Handmade in any of our 30-plus flavors or best selling variety assortments like Fabulous Four. This cake serves eight with tasty 4 oz servings. Available in 32 oz - 6 to a case pack Petite Retail Cheesecake This dessert comes in a convenient box that allows multiple flavors to a pack. A great holiday item! Grab-n-Go appetizer cheesecakes for parties. -
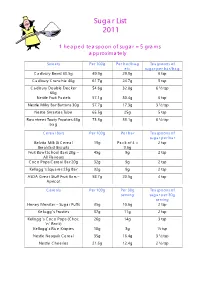
Sugar List 2011
Sugar List 2011 1 heaped teaspoon of sugar = 5 grams approximately Sweets Per 100g Per bar/bag Teaspoons of etc sugar per bar/bag Cadbury Boost 60.5g 49.9g 29.9g 6 tsp Cadbury Crunchie 40g 61.7g 24.7g 5 tsp Cadbury Double Decker 54.6g 32.8g 6 ½ tsp 60g Nestle Fruit Pastels 57.1g 30.4g 6 tsp Nestle Milky Bar Buttons 30g 57.7g 17.3g 3 ½ tsp Nestle Smarties Tube 65.5g 25g 5 tsp Rowntrees Tooty Frooties 48g 73.3g 33.1g 6 ½ tsp bag Cereal Bars Per 100g Per bar Teaspoons of sugar per bar Belvita Milk & Cereal 19g Pack of 4 = 2 tsp Breakfast Biscuits 9.6g Fruit Bowl School Bars 20g – 45g 9g 2 tsp All Flavours Coco Pops Cereal Bar 20g 32g 9g 2 tsp Kellogg’s Squares 25g Bar 32g 9g 2 tsp ASDA Great Stuff Fruit Bars – 58.7g 20.5g 4 tsp Apricot Cereals Per 100g Per 30g Teaspoons of serving sugar per 30g serving Honey Monster – Sugar Puffs 35g 10.6g 2 tsp Kellogg’s Frosties 37g 11g 2 tsp Kellogg’s Coco Pops (Choc 26g 14g 3 tsp ‘n’ Rock) Kellogg’s Rice Krispies 10g 3g ½ tsp Nestle Nesquik Cereal 35g 16.4g 3 ½ tsp Nestle Cheerios 21.6g 12.4g 2 ½ tsp Biscuits Per 100g Per biscuit Teaspoons of sugar per biscuit Burton’s Jammie Dodgers 36.2g 6.8g 1 ½ tsp Fox’s Mini Malted Moos 19.5g 4.9g 1 tsp Nestle Kitkat (2 finger bar) 47.8g 10g 2 tsp Oreo Cookies 40.5g 4.5g 1tsp Rocky Caramel Biscuit Bar 49.2g 10.4g 2 tsp Tunnocks Milk Chocolate 36g 8.6g 2 tsp Tea Cakes Yoghurt & Dessert Per 100g Per pot Teaspoons of sugar per pot Ambrosia Devon Custard 12g 18g 4 tsp Pot (150g) Muller Little Stars Fruit Juice 11.7g 8.8g 2 tsp Jelly 75g Muller Little Stars Fromage 11.7g -
Welcome to Luscious.™
WOMEN’S BUSINESS ENTERPRISE. SWEET STREET DESSERTS IS A CERTIFIED Welcome to luscious.™ 722 Hiesters Lan Welcome to luscious. e , r e a d i n ™ g , www.sweetstreet.com P P: 1 800 793 3897 F: 1 610 921 0915 a 1 9 6 0 5 SWEET STREET DESSERTS ARE KOShER DAIRy(KD) WITh ThE ExCEPTION OF ChEWy MARShMAllOW SqUARES, ChOCOlATE MARShMAllOW BAR, EUROPEAN INDIvIDUAlS, GRAND CRU™ PETIT FOURS AND SCONES. ©2010, SWEET STREET DESSERTS. All RIGhTS RESERvED. PRINTED IN ThE U.S.A. Oi 4/10 50M Welcome to SWeet Street DeSSertS Dedication to Quality 1–3 100% Guarantee 3 WE HAVE A Important customers 4 leVerAGING lUScIoUS 5–9 WE AT SWEET STREET DESSERTS ™ MISSION Balancing Desserts 6, 7 CHOOSE TO BE THE LEADER IN THE Support Programs 8, 9 Artisan Scone Variety cups & caddies 10 GOURMET DESSERT INDUSTRY, SETTING THE Units POrtiOn POrtiOn POrtiOn Sweet elite Program 11 COde Per Case STANDARD Per Unit Per CBYase WHICHWt ALL OTHERS ARE JUDGED. 7968 4 13 52 3.5 oz 2 blueberry scones PIeS 12 twelve-Inch 12, 13 Deep Dish 14, 15 eing a leader in “sweet indulgence” is our way 1 cinnamon scone Petal Pan 15, 16 1 chocolate chip scone Variety Packs 17 of life. To maintain our leadership, we will be tHe BIG lINe 18 Pies 18 B missionaries of the dessert experience, expanding its cakes 19–21 pleasures to all parts of the day and all parts of the world. cAKeS 22 Nine-Inch round 22–24 With our unwavering commitment to profitably create a mousse 24 constantly2 chocolate evolving chunk cookies and extraordinary line of great-tasting, tiramisu 25 1 oatmeal raisin cookie Variety Packs 25 high quality desserts, innovation will always be essential cHeeSecAKeS 26 1 butter sugar cookie Nine-Inch 26–28 to our success and cutting-edge products will always ten-Inch 28 be our future.