Pharmacological Chaperone Action in Humanized Mouse Models of MC4R-Linked Obesity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Preclinical Effects of Melanocortins in Male Sexual Dysfunction
International Journal of Impotence Research (2008) 20, S11–S16 & 2008 Nature Publishing Group All rights reserved 0955-9930/08 $30.00 www.nature.com/ijir Preclinical effects of melanocortins in male sexual dysfunction AM Shadiack1 and S Althof2 1Locus Pharmaceuticals, Blue Bell, PA, USA and 2The Center for Marital and Sexual Health of South Florida, West Palm Beach, FL, USA The neurobiology of sexual behavior involves the interrelationships between sex steroids and neurotransmitters that result in both central nervous system (CNS) effects and effects in the genitalia. Tools such as positron emission tomography (PET) and functional magnetic resonance imaging (fMRI) scanning can help determine what areas of the brain are activated under sexual stimulation. Our understanding of the role of various neurotransmitters, neurosteroids and other CNS-acting compounds is improving. The role of CNS-acting compounds such as dopamine agonists in the treatment of male sexual dysfunction is under active investigation. Melanocortins have CNS and peripheral roles in a wide variety of bodily functions. The melanocortin agonist bremelanotide appears to act in the CNS to promote erections in preclinical models, and may also stimulate behaviors that facilitate sexual activity beyond their erectogenic effects. International Journal of Impotence Research (2008) 20, S11–S16; doi:10.1038/ijir.2008.17 Keywords: erectile dysfunction; neuroanatomy; neurophysiology; CNS-acting agents; melanocortins; bremelanotide Introduction Neuroanatomy of male sexual response The neurobiology of sexual behavior involves the Sex and the brain interrelationships between sex steroids and neuro- Sexual arousal can now be studied with such transmitters that result in both central nervous sophisticated tools as PET (positron emission system (CNS) effects and effects in the genitalia. -
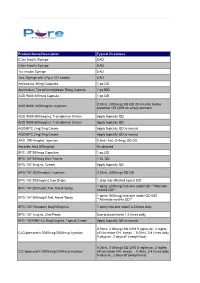
Pure Pricing List
Product Name/Description Typical Directions 0.3cc Insulin Syringe UAD 0.5cc Insulin Syringe UAD 1cc Insulin Syringe UAD 1mL Syringe with 27g x 1/2" needle UAD Amlexanox 40mg Capsules 1 po QD Ammonium Tetrathiomolybdate 50mg Capsule 1 po BID AOD 9604 600mcg Capsule 1 po QD 0.25mL (300mcg) SQ QD 30 minutes before AOD 9604 1200mcg/mL Injection breakfast OR QHS on empty stomach AOD 9604 600mcg/mL Transdermal Cream Apply topically QD AOD 9604 600mcg/mL Transdermal Cream Apply topically QD AOD/BPC 2mg/2mg Cream Apply topically QD to wound AOD/BPC 2mg/2mg Cream Apply topically QD to wound ARA 290 4mg/mL Injection 0.5mL-1mL (2-4mg) SQ QD Ascorbic Acid 500mg/mL As directed BPC-157 500mcg Capsules 1 po QD BPC-157 500mcg Mini-Troche 1 SL QD BPC-157 2mg/mL Cream Apply topically QD BPC-157 2000mcg/mL Injection 0.25mL (500mcg) SQ QD BPC-157 200mcg/mL Eye Drops 1 drop into affected eye(s) BID 1 spray (200mcg) into one nostril QD **Alternate BPC-157 200mcg/0.1mL Nasal Spray nostrils QD** 1 spray (500mcg) into one nostril QD-BID BPC-157 500mcg/0.1mL Nasal Spray **Alternate nostrils QD** BPC-157/ Synapsin 5mg/50mg/mL 1 spray into one nostril 2-3 times daily BPC-157 1mg/mL Oral Paste Use to brush teeth 1-2 times daily BPC-157/GHK-Cu 2mg/2mg/mL Topical Cream Apply topically QD to wound 0.05mL (100mcg) SQ QHS 5 nights on, 2 nights CJC/Ipamorelin 2000mcg/2000mcg Injection off (Increase GH, sleep)… 0.05mL 3-4 times daily 5 days on, 2 days off (weight loss) 0.05mL (100mcg) SQ QHS 5 nights on, 2 nights CJC/Ipamorelin 2000mcg/2000mcg Injection off (Increase GH, -

Zebrafish Bioassay for Screening Therapeutic Candidates Based On
International Journal of Molecular Sciences Article Zebrafish Bioassay for Screening Therapeutic Candidates Based on Melanotrophic Activity Ted I. Hong 1,†, Kyu-Seok Hwang 2,† , Tae-Ik Choi 1, Gunnar Kleinau 3, Patrick Scheerer 3 , Jeong Kyu Bang 4 , Seung-Hyun Jung 5,* and Cheol-Hee Kim 1,* 1 Department of Biology, Chungnam National University, Daejeon 34134, Korea; [email protected] (T.I.H.); [email protected] (T.-I.C.) 2 Drug Discovery Platform Research Center, Korea Research Institute of Chemical Technology, Daejeon 34114, Korea; [email protected] 3 Group Protein X-ray Crystallography and Signal Transduction, Institute of Medical Physics and Biophysics, Charité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, D-10117 Berlin, Germany; [email protected] (G.K.); [email protected] (P.S.) 4 Division of Magnetic Resonance, Korea Basic Science Institute, Ochang, Cheongju 28119, Korea; [email protected] 5 Department of Applied Marine Bioresource Science, National Marine Biodiversity Institute of Korea, Seocheon 33662, Korea * Correspondence: [email protected] (S.-H.J.); [email protected] (C.-H.K.) † These authors contributed equally to this work. Abstract: In this study, we used the zebrafish animal model to establish a bioassay by which physi- ological efficacy differential of alpha-melanocyte-stimulating hormone (α-MSH) analogues could be measured by melanosome dispersion in zebrafish larvae. Brain-skin connection research has Citation: Hong, T.I.; Hwang, K.-S.; purported the interconnectedness between the nervous system and skin physiology. Accordingly, the Choi, T.-I.; Kleinau, G.; Scheerer, P.; neuropeptide α-MSH is a key regulator in several physiological processes, such as skin pigmentation Bang, J.K.; Jung, S.-H.; Kim, C.-H. -

Targeting Central Melanocortin Receptors: a Promising Novel Approach for Treating Alcohol Abuse Disorders
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Frontiers - Publisher Connector REVIEW ARTICLE published: 03 June 2014 doi: 10.3389/fnins.2014.00128 Targeting central melanocortin receptors: a promising novel approach for treating alcohol abuse disorders Jeffrey J. Olney 1, Montserrat Navarro 1 and Todd E. Thiele 1,2* 1 Department of Psychology, University of North Carolina, Chapel Hill, NC, USA 2 Bowles Center for Alcohol Studies, University of North Carolina, Chapel Hill, NC, USA Edited by: The melanocortin (MC) peptides are produced centrally by propiomelanocortin (POMC) Richard Lowell Bell, Indiana neurons within the arcuate nucleus of the hypothalamus and act through five University School of Medicine, USA seven-transmembrane G-protein coupled melanocortin receptor (MCR) subtypes. The Reviewed by: MC3R and MC4R subtypes, the most abundant central MCRs, are widely expressed Gregg E. Homanics, University of Pittsburgh, USA in brain regions known to modulate neurobiological responses to ethanol, including Luigia Trabace, University of Foggia, regions of the hypothalamus and extended amygdala. Agouti-related protein (AgRP), Italy also produced in the arcuate nucleus, is secreted in terminals expressing MCRs and Olivier George, The Scripps functions as an endogenous MCR antagonist. This review highlights recent genetic and Research Institute, USA pharmacological findings that have implicated roles for the MC and AgRP systems in *Correspondence: Todd E. Thiele, Department of modulating ethanol consumption. Ethanol consumption is associated with significant Psychology, University of North alterations in the expression levels of various MC peptides/protein, which suggests that Carolina, Davie Hall, CB #3270, ethanol-induced perturbations of MC/AgRP signaling may modulate excessive ethanol Chapel Hill, NC 27599-3270, USA intake. -

Professional Monograph Melanotan II
Professional Monograph Melanotan II SUMMARY ● Meltanotan II - synthetic alpha melanocyte stimulating hormone (α -MSH) ● Non-specific central melanocortin receptor agonist – MC1R, MC3R, MC4R, MC5R ● Melanotan I (afamelanotide) has the same mechanism of action as Melantotan II, however Melanotan I doesn’t cross the BBB so no central effects (the metabolism support and sexual stimulation) like Melanotan II ● Supports melanogenesis ● Photoprotective ● Improves libido ● Immune – activates Treg cells and improves Th1/Th17 balance ● Reduces appetite and improves lipid and glucose regulation ● Neuroprotective – decreases neuroinflammation © Copyright 2018, Scientific Performance Research, LLC" Cincinnati, OH ● Dopamine modulating Uses ● Tanning ● Protection from phototoxicity o Erythropoietic protoporphyria ● Metabolic Support o Lipolytic, appetite control, anti-inflammatory, decreases oxidative stress ● Conditions requiring immune balance o Autoimmune conditions ● Libido enhancement ● Potential use in alcohol and drug abuse, including opioids, cocaine (reward cascade supportive) DOSAGE ● General dosage o Tanning ▪ 200 mcg SubQ daily for 1 week ▪ Adjust according to pigment changes ▪ After pigment stabilizes, 100 mcg SC 2 x a week o Metabolic Enhancement ▪ 50 mcg daily SubQ and adjust o Immunity ▪ 200 mcg daily SubQ for 6-8 weeks ▪ Use until clinical effects are pronounced o Sexual stimulation ▪ More of a Side effect so occurs with any dosage regimen ▪ Higher doses (ie 500 – 1,000 mcg) can lead to more pronounced stimulation ▪ Melanotan sexual -

Resolution Pharmacology Therapeutic Innovation in Inflammation.ACTH: the Forgotten Therapy Trinidad Montero-Melendez the William
Resolution Pharmacology therapeutic innovation in inflammation.ACTH: The Forgotten Therapy Trinidad Montero-Melendez The William Harvey Research Institute, Barts and The London School of Medicine, Queen Mary University of London. Charterhouse Square, EC1M 6BQ, London, United Kingdom. Correspondence: Trinidad Montero-Melendez, PhD Address: The William Harvey Research Institute, Barts and The London School of Medicine, Charterhouse Square, EC1M 6BQ, London, United Kingdom. Telephone: +44-207-8825654 Fax: +44-207-8826076 E-mail: [email protected] Abstract Although anti-inflammatory drugs are among the most common class of marketed drugs, chronic inflammatory conditions such as rheumatoid arthritis, multiple sclerosis or inflammatory bowel disease still represent unmet needs. New first-in-class drugs might be discovered in the future but the repurpose and further development of old drugs also offers promise for these conditions. This is the case of the melanocortin adrenocorticotropin hormone, ACTH, used in patients since 1952 but regarded as the last therapeutic option when other medications, such as glucocorticoids, cannot be used. Better understanding on its physiological and pharmacological mechanisms of actions and new insights on melanocortin receptors biology have revived the interest on rescuing this old and effective drug. ACTH does not only induce cortisol production, as previously assumed, but it also exerts anti-inflammatory actions by targeting melanocortin receptors present on immune cells. The endogenous agonists for these receptors (ACTH, α-, β-, and γ-melanocyte stimulating hormones), are also produced locally by immune cells, indicating the existence of an endogenous anti-inflammatory tissue-protective circuit involving the melanocortin system. These findings suggested that new ACTH-like melanocortin drugs devoid of steroidogenic actions, and hence side effects, could be developed. -

Unabated Anorexic and Enhanced Thermogenic Responses to Melanotan II in Diet-Induced Obese Rats Despite Reduced Melanocortin 3 and 4 Receptor Expression
123 Unabated anorexic and enhanced thermogenic responses to melanotan II in diet-induced obese rats despite reduced melanocortin 3 and 4 receptor expression GLi1, Y Zhang1,2, J T Wilsey1,2 and P J Scarpace1,2 1Department of Pharmacology and Therapeutics, University of Florida College of Medicine, Gainesville, Florida 32610, USA 2Geriatric Research, Education and Clinical Center (182), Department of Veterans Affairs Medical Center, Gainesville, Florida 32608-1197, USA (Requests for offprints should be addressed to P J Scarpace, Geriatric Research, Education and Clinical Center, Malcolm Randall Veterans Affairs Medical Center, Gainesville, Florida 32608-1197, USA; Email: scarpace@ufl.edu) Abstract The effects of the chronic activation of the central melano- expression and prevented the reduction in muscle-type cortin (MC) system by melanotan II (MTII) were assessed carnitine palmitoyltransferase I mRNA by pair-feeding in in chow-fed (CH) and high-fat (HF) diet-induced obese the muscle of DIO rats. Compared with CH controls, (DIO) Sprague–Dawley rats. Six-day central infusion of hypothalamic MC3 and MC4 receptor expression levels MTII (1 nmol/day) reduced body weight and visceral were reduced in DIO controls. This study has demon- adiposity compared with ad libitum-fed control and pair- strated that, despite reduced hypothalamic MC3/MC4 fed groups and markedly suppressed caloric intake in both receptor expression, anorexic and thermogenic responses CH and DIO rats. The anorexic response to MTII was to MTII are unabated with an initial augmentation of similar in DIO relative to CH rats. MTII induced a energy expenditure in DIO versus CH rats. The HF- sustained increase in oxygen consumption in DIO but a induced up-regulation of UCP1 in BAT may contribute delayed response in CH rats. -

Supplementary Data
Supplementary data Table S1. Results of the docking of DrugBank compounds onto GRP78 (NBD) (only molecules with lower docking scores than control ATP are presented). Molecule name Final docking score S (kcal/mol) Imatinib -9.26206 FK-614 -8.9803 Selonsertib -8.85865 Sorafenib -8.841712 CID 5288250 -8.6442 Pemetrexed -8.6247129 4SC-203 -8.61179 Zafirlukast -8.59865 (2S)-2-[[4-[2-[(6S)-2-Amino-4-oxo-5,6,7,8-tetrahydro-3H-pyrido[2,3-d]pyrimidin- -8.58657 6-yl]ethyl]benzoyl]amino]pentanedioic acid Icariin -8.46474 Raltegravir -8.44994 4-[(5-{[4-(3-Chlorophenyl)-3-oxopiperazin-1-YL]methyl}-1H-imidazol-1- -8.44899 YL)methyl]benzonitrile Dacomitinib -8.4483 [(1S)-1-Cyclohexyloxycarbonyloxyethyl] 2-ethoxy-3-[[4-[2-(2H-tetrazol-5- -8.43484 yl)phenyl]phenyl]methyl]benzimidazole-4-carboxylate Darexaban -8.4267 Tenofovir disoproxil -8.4124937 Neratinib -8.39573 Ponatinib -8.38035 6-(3-(Dimethylcarbamoyl)phenylsulfonyl)-4-(3-methoxyphenylamino)-8- -8.32773 methylquinoline-3-carboxamide 2'-Deoxy-N-(naphthalen-1-ylmethyl)guanosine 5'-(dihydrogen phosphate) -8.31957 3-(5-{[4-(Aminomethyl)piperidin-1-Yl]methyl}-1h-Indol-2-Yl)quinolin-2(1h)-One -8.30333 (2R)-N-[2-[4-[5-[4-[(4-Acetamidophenyl)methoxy]-2,3-dichlorophenyl]-2- -8.2941 methylpyrazol-3-yl]piperidin-1-yl]-2-oxoethyl]-2-(diaminomethylideneamino)- 4-methylpentanamide Butafenacil -8.29337 Nilotinib -8.28767 Gedatolisib -8.27715 N-(Sulfanylacetyl)tyrosylprolylmethioninamide -8.27166 Leucovorin -8.2563314 Asp3026 -8.24956 Methyl (1R,2S)-2-(hydroxycarbamoyl)-1-[[4-[(2-methylquinolin-4- -8.2435 yl)methoxy]phenyl]methyl]cyclopropane-1-carboxylate -

Melanocortin Receptor Agonists Facilitate Oxytocin-Dependent Partner Preference Formation in the Prairie Vole
Neuropsychopharmacology (2015) 40, 1856–1865 © 2015 American College of Neuropsychopharmacology. All rights reserved 0893-133X/15 www.neuropsychopharmacology.org Melanocortin Receptor Agonists Facilitate Oxytocin-Dependent Partner Preference Formation in the Prairie Vole *,1,2 1 1 1 2,3 Meera E Modi , Kiyoshi Inoue , Catherine E Barrett , Kara A Kittelberger , Daniel G Smith , 4 1 Rainer Landgraf and Larry J Young 1 Center Translational Social Neuroscience, Silvio O Conte Center for Oxytocin and Social Cognition, Department of Psychiatry and Behavioral 2 Sciences, Yerkes National Primate Research Center, Emory University, Atlanta, GA, USA; Neuroscience Research Unit, Pfizer Worldwide Research 3 4 and Development, Cambridge, MA, USA; Autism Speaks, Boston, MA, USA; Max Plank Institute of Psychiatry, Munich, Germany The central melanocortin (MC) system has been widely studied for its effects on food intake and sexual behavior. However, the MC system, and more specifically the MC4 receptor (MC4R), also interacts with neurochemical systems that regulate socioemotional behaviors, including oxytocin (OT) and dopamine. In monogamous prairie voles, OT and dopamine interact to promote partner preference formation, a laboratory measure of an enduring social bond between mates. Here we investigated the effects of MC receptor activation on partner preference formation in prairie voles, as well as the interaction between the MC and OT systems during this process. Peripheral administration of the brain penetrant MC3/4R receptor peptide agonist, Melanotan II (MTII), and the highly selective, small- molecule MC4R agonist, Pf-446687, enhanced partner preference formation in the prairie vole, but not in the non-monogamous meadow vole. MTII-induced partner preferences were enduring, as they were present 1 week after drug manipulation. -

Oxytocin Neurone Activity and Release Following Administration of Melanotan-II in Anaesthetised Rats
Oxytocin neurone activity and release following administration of Melanotan-II in anaesthetised rats Luis Paiva Doctor of Philosophy The University of Edinburgh 2017 Declaration I declare that this thesis has been composed by myself, and that the experiments described here were performed by myself at the Centre for Integrative Physiology, University of Edinburgh, and that any technical contribution made by others have been clearly indicated. I also declare that this work has not been submitted for any other degree or professional qualification. Luis Paiva July 2017 ii Abstract Oxytocin release within the brain modulates several social behaviours in animals and humans. Moreover, low central oxytocin content has been linked to neuropsychiatric disorders, such as anxiety and autism. The exogenous administration of oxytocin has been proposed for therapeutic treatment, but oxytocin does not cross the blood-brain barrier (BBB) in physiologically significant amounts. An alternative approach to oxytocin administration is to stimulate central oxytocin release using melanocortins. Central administration of the naturally occurring melanocortin, α-MSH, has been shown to trigger somatodendritic oxytocin release in vitro. Unfortunately, endogenous melanocortins also do not penetrate the BBB in neuroactive amounts. In this study, I investigated whether systemic administration of synthetic melanocortin receptor 3/4 (MC3/4) agonist, Melanotan-II (MT-II), affects oxytocin neuronal activity and secretion in anaesthetised rats. I hypothesised that systemic -

In Vivo Evidence for Inverse Agonism of Agouti-Related Peptide in the Central Nervous System of Proopiomelanocortin-Deficient Mice Virginie Tolle1,2 and Malcolm J
ORIGINAL ARTICLE In Vivo Evidence for Inverse Agonism of Agouti-Related Peptide in the Central Nervous System of Proopiomelanocortin-Deficient Mice Virginie Tolle1,2 and Malcolm J. Low1,2,3 OBJECTIVE—Melanocyte-stimulating hormone (MSH) pep- tides processed from proopiomelanocortin (POMC) regulate energy homeostasis by activating neuronal melanocortin recep- enetic disruption of either mouse or human tor (MC-R) signaling. Agouti-related peptide (AgRP) is a naturally proopiomelanocortin (POMC) causes early-on- occurring MC-R antagonist but also displays inverse agonism at set obesity (1–3), highlighting a major role of constitutively active melanocortin-4 receptor (MC4-R) expressed POMC in the regulation of energy homeostasis. on transfected cells. We investigated whether AgRP functions G POMC is processed posttranslationally into multiple pep- similarly in vivo using mouse models that lack all neuronal MSH,  thereby precluding competitive antagonism of MC-R by AgRP. tides, including the opioid -endorphin and the melano- cortins ACTH, ␣-melanocyte-stimulating hormone (␣MSH), RESEARCH DESIGN AND METHODS—Feeding and meta-  ␥ bolic effects of the MC-R agonist melanotan II (MTII), AgRP, and MSH, and MSH. POMC peptides in the central nervous ghrelin were investigated after intracerebroventricular injection system (CNS) are essential in the regulation of energy Ϫ/Ϫ ϩ intake and expenditure as demonstrated in studies using in neural-specific POMC-deficient (Pomc Tg/ ) and global Ϫ Ϫ POMC-deficient (PomcϪ/Ϫ) mice. Gene expression was quanti- compound mutant mice (Pomc / Tg/ϩ) expressing a fied by RT-PCR. Pomc transgene that selectively restored pituitary POMC Ϫ/Ϫ RESULTS—Hyperphagic POMC-deficient mice were more sen- in Pomc mice to produce a neural-selective POMC sitive than wild-type mice to the anorectic effects of MTII. -

Melanocortin Receptor Agonists, Penile Erection, and Sexual Motivation: Human Studies with Melanotan II
International Journal of Impotence Research (2000) 12, Suppl 4, S74±S79 ß 2000 Macmillan Publishers Ltd All rights reserved 0955-9930/00 $15.00 www.nature.com/ijir Melanocortin receptor agonists, penile erection, and sexual motivation: human studies with Melanotan II H Wessells1*, N Levine1, ME Hadley2, R Dorr3 and V Hruby4 1Sections of Urology and Dermatology, The University of Arizona College of Medicine, and Departments of 2Cell Biology, 3Pharmacology and 4Chemistry, The University of Arizona, Tucson, AZ, USA We review our experience with Melanotan II, a non-selective melanocortin receptor agonist, in human subjects with erectile dysfunction (ED). Melanotan II was administered to 20 men with psychogenic and organic ED using a double-blind placebo-controlled crossover design. Penile rigidity was monitored for 6 h using RigiScan. Level of sexual desire and side effects were reported with a questionnaire. In the absence of sexual stimulation, Melanotan II led to penile erection in 17 of 20 men. Subjects experienced a mean of 41 min Rigiscan tip rigidity > 80%. Increased sexual desire was reported after 13=19 (68%) doses of Melanotan II vs 4=21 (19%) of placebo (P < 0.01). Nausea and yawning were frequently reported side effects due to Melanotan II; at a dose of 0.025 mg=kg, 12.9% of subjects had severe nausea. We conclude that Melanotan II is a potent initiator of penile erection in men with erectile dysfunction. Our ®ndings warrant further investigation of melanocortin agonists and antagonists on penile erection. International Journal