Regulation of Skin Barrier Function Via Competition Between AHR Axis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

18 December 2020 – to Date)
(18 December 2020 – to date) MEDICINES AND RELATED SUBSTANCES ACT 101 OF 1965 (Gazette No. 1171, Notice No. 1002 dated 7 July 1965. Commencement date: 1 April 1966 [Proc. No. 94, Gazette No. 1413] SCHEDULES Government Notice 935 in Government Gazette 31387 dated 5 September 2008. Commencement date: 5 September 2008. As amended by: Government Notice R1230 in Government Gazette 32838 dated 31 December 2009. Commencement date: 31 December 2009. Government Notice R227 in Government Gazette 35149 dated 15 March 2012. Commencement date: 15 March 2012. Government Notice R674 in Government Gazette 36827 dated 13 September 2013. Commencement date: 13 September 2013. Government Notice R690 in Government Gazette 36850 dated 20 September 2013. Commencement date: 20 September 2013. Government Notice R104 in Government Gazette 37318 dated 11 February 2014. Commencement date: 11 February 2014. Government Notice R352 in Government Gazette 37622 dated 8 May 2014. Commencement date: 8 May 2014. Government Notice R234 in Government Gazette 38586 dated 20 March 2015. Commencement date: 20 March 2015. Government Notice 254 in Government Gazette 39815 dated 15 March 2016. Commencement date: 15 March 2016. Government Notice 620 in Government Gazette 40041 dated 3 June 2016. Commencement date: 3 June 2016. Prepared by: Page 2 of 199 Government Notice 748 in Government Gazette 41009 dated 28 July 2017. Commencement date: 28 July 2017. Government Notice 1261 in Government Gazette 41256 dated 17 November 2017. Commencement date: 17 November 2017. Government Notice R1098 in Government Gazette 41971 dated 12 October 2018. Commencement date: 12 October 2018. Government Notice R1262 in Government Gazette 42052 dated 23 November 2018. -

Cytokine Nomenclature
RayBiotech, Inc. The protein array pioneer company Cytokine Nomenclature Cytokine Name Official Full Name Genbank Related Names Symbol 4-1BB TNFRSF Tumor necrosis factor NP_001552 CD137, ILA, 4-1BB ligand receptor 9 receptor superfamily .2. member 9 6Ckine CCL21 6-Cysteine Chemokine NM_002989 Small-inducible cytokine A21, Beta chemokine exodus-2, Secondary lymphoid-tissue chemokine, SLC, SCYA21 ACE ACE Angiotensin-converting NP_000780 CD143, DCP, DCP1 enzyme .1. NP_690043 .1. ACE-2 ACE2 Angiotensin-converting NP_068576 ACE-related carboxypeptidase, enzyme 2 .1 Angiotensin-converting enzyme homolog ACTH ACTH Adrenocorticotropic NP_000930 POMC, Pro-opiomelanocortin, hormone .1. Corticotropin-lipotropin, NPP, NP_001030 Melanotropin gamma, Gamma- 333.1 MSH, Potential peptide, Corticotropin, Melanotropin alpha, Alpha-MSH, Corticotropin-like intermediary peptide, CLIP, Lipotropin beta, Beta-LPH, Lipotropin gamma, Gamma-LPH, Melanotropin beta, Beta-MSH, Beta-endorphin, Met-enkephalin ACTHR ACTHR Adrenocorticotropic NP_000520 Melanocortin receptor 2, MC2-R hormone receptor .1 Activin A INHBA Activin A NM_002192 Activin beta-A chain, Erythroid differentiation protein, EDF, INHBA Activin B INHBB Activin B NM_002193 Inhibin beta B chain, Activin beta-B chain Activin C INHBC Activin C NM005538 Inhibin, beta C Activin RIA ACVR1 Activin receptor type-1 NM_001105 Activin receptor type I, ACTR-I, Serine/threonine-protein kinase receptor R1, SKR1, Activin receptor-like kinase 2, ALK-2, TGF-B superfamily receptor type I, TSR-I, ACVRLK2 Activin RIB ACVR1B -

Classification Decisions Taken by the Harmonized System Committee from the 47Th to 60Th Sessions (2011
CLASSIFICATION DECISIONS TAKEN BY THE HARMONIZED SYSTEM COMMITTEE FROM THE 47TH TO 60TH SESSIONS (2011 - 2018) WORLD CUSTOMS ORGANIZATION Rue du Marché 30 B-1210 Brussels Belgium November 2011 Copyright © 2011 World Customs Organization. All rights reserved. Requests and inquiries concerning translation, reproduction and adaptation rights should be addressed to [email protected]. D/2011/0448/25 The following list contains the classification decisions (other than those subject to a reservation) taken by the Harmonized System Committee ( 47th Session – March 2011) on specific products, together with their related Harmonized System code numbers and, in certain cases, the classification rationale. Advice Parties seeking to import or export merchandise covered by a decision are advised to verify the implementation of the decision by the importing or exporting country, as the case may be. HS codes Classification No Product description Classification considered rationale 1. Preparation, in the form of a powder, consisting of 92 % sugar, 6 % 2106.90 GRIs 1 and 6 black currant powder, anticaking agent, citric acid and black currant flavouring, put up for retail sale in 32-gram sachets, intended to be consumed as a beverage after mixing with hot water. 2. Vanutide cridificar (INN List 100). 3002.20 3. Certain INN products. Chapters 28, 29 (See “INN List 101” at the end of this publication.) and 30 4. Certain INN products. Chapters 13, 29 (See “INN List 102” at the end of this publication.) and 30 5. Certain INN products. Chapters 28, 29, (See “INN List 103” at the end of this publication.) 30, 35 and 39 6. Re-classification of INN products. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
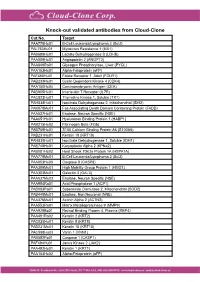
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1) -

Evolutionary Divergence and Functions of the Human Interleukin (IL) Gene Family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W
UPDATE ON GENE COMPLETIONS AND ANNOTATIONS Evolutionary divergence and functions of the human interleukin (IL) gene family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W. Nebert3* and Vasilis Vasiliou1 1Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences, University of Colorado Denver, Aurora, CO 80045, USA 2Department of Clinical Pharmacy, University of Colorado Denver, Aurora, CO 80045, USA 3Department of Environmental Health and Center for Environmental Genetics (CEG), University of Cincinnati Medical Center, Cincinnati, OH 45267–0056, USA *Correspondence to: Tel: þ1 513 821 4664; Fax: þ1 513 558 0925; E-mail: [email protected]; [email protected] Date received (in revised form): 22nd September 2010 Abstract Cytokines play a very important role in nearly all aspects of inflammation and immunity. The term ‘interleukin’ (IL) has been used to describe a group of cytokines with complex immunomodulatory functions — including cell proliferation, maturation, migration and adhesion. These cytokines also play an important role in immune cell differentiation and activation. Determining the exact function of a particular cytokine is complicated by the influence of the producing cell type, the responding cell type and the phase of the immune response. ILs can also have pro- and anti-inflammatory effects, further complicating their characterisation. These molecules are under constant pressure to evolve due to continual competition between the host’s immune system and infecting organisms; as such, ILs have undergone significant evolution. This has resulted in little amino acid conservation between orthologous proteins, which further complicates the gene family organisation. Within the literature there are a number of overlapping nomenclature and classification systems derived from biological function, receptor-binding properties and originating cell type. -

Np63 Regulates IL-33 and IL-31 Signaling in Atopic Dermatitis
Cell Death and Differentiation (2016) 23, 1073–1085 OPEN & 2016 Macmillan Publishers Limited All rights reserved 1350-9047/16 www.nature.com/cdd ΔNp63 regulates IL-33 and IL-31 signaling in atopic dermatitis JM Rizzo1,2, A Oyelakin3, S Min3, K Smalley1, J Bard1, W Luo1,7, J Nyquist4, E Guttman-Yassky5, T Yoshida6, A De Benedetto6, LA Beck6, S Sinha*,1 and R-A Romano*,3 Atopic dermatitis (AD) is the most common inflammatory skin disease with no well-delineated cause or effective cure. Here we show that the p53 family member p63, specifically the ΔNp63, isoform has a key role in driving keratinocyte activation in AD. We find that overexpression of ΔNp63 in transgenic mouse epidermis results in a severe skin phenotype that shares many of the key clinical, histological and molecular features associated with human AD. This includes pruritus, epidermal hyperplasia, aberrant keratinocyte differentiation, enhanced expression of selected cytokines and chemokines and the infiltration of large numbers of inflammatory cells including type 2 T-helper cells – features that are highly representative of AD dermatopathology. We further demonstrate several of these mediators to be direct transcriptional targets of ΔNp63 in keratinocytes. Of particular significance are two p63 target genes, IL-31 and IL-33, both of which are key players in the signaling pathways implicated in AD. Importantly, we find these observations to be in good agreement with elevated levels of ΔNp63 in skin lesions of human patients with AD. Our studies reveal an important role for ΔNp63 in the pathogenesis of AD and offer new insights into its etiology and possible therapeutic targets. -

Atopic Dermatitis in Animals and People: an Update and Comparative Review
veterinary sciences Review Atopic Dermatitis in Animals and People: An Update and Comparative Review Rosanna Marsella 1,2,* and Anna De Benedetto 1 1 Department of Dermatology, College of Medicine, University of Florida, 4037 NW 86 Terrace, Gainesville, FL 32606, USA; [email protected]fl.edu 2 Department of Small Animal Clinical Sciences, College of Veterinary Medicine, University of Florida, 2015 SW 16th Avenue, Gainesville, FL 32610, USA * Correspondence: Marsella@ufl.edu; Tel.: +1-352-278-0742 Academic Editor: Rob Siebers Received: 13 June 2017; Accepted: 22 July 2017; Published: 26 July 2017 Abstract: Atopic dermatitis is an extremely common, pruritic, and frustrating disease to treat in both people and animals. Atopic dermatitis is multifactorial and results from complex interactions between genetic and environmental factors. Much progress has been done in recent years in terms of understanding the complex pathogenesis of this clinical syndrome and the identification of new treatments. As we learn more about it, we appreciate the striking similarities that exist in the clinical manifestations of this disease across species. Both in animals and people, atopic disease is becoming increasingly common and important similarities exist in terms of immunologic aberrations and the propensity for allergic sensitization. The purpose of this review is to highlight the most recent views on atopic dermatitis in both domestic species and in people emphasizing the similarities and the differences. A comparative approach can be beneficial in understanding the natural course of this disease and the variable response to existing therapies. Keywords: atopic dermatitis; skin barrier; atopic march; dogs; cats; horses; humans 1. Introduction Increasing awareness exists with respect to the similarities between animal and human diseases and the appreciation of how a comparative approach is useful to improve our understanding of pathogenesis and the development of new treatments [1,2]. -

World Congress Veterinary Dermatology (WCVD) Summary by Dr Carmen Lorente, DVM, Phd, Dipecvd EBVS® European Specialist in Veterinary Dermatology
Quality Veterinary Diagnostics BattLab from disease to optimal health Quality Veterinary Diagnostics from Disease to Optimal Health LABORATORY FOR CLINICAL DIAGNOSTICS Third World Congress Veterinary Dermatology (WCVD) Summary By Dr Carmen Lorente, DVM, PhD, DipECVD EBVS® European Specialist in Veterinary Dermatology Our third summary from the digital World Congress in Veterinary Dermatology 2020 (WCVD) focuses on two exciting therapies: biological therapy and bacteriophage therapy. They are old therapies but with a tremendous and developing future in managing complex diseases including resistant bacterial, atopic dermatitis, pruritus and cancer. Below are some summary points from the talk by Dr Douglas DeBoer on the subject of “Biological therapies in dermatology” and the lecture “Bacteriophage therapy for challenging bacterial infections” from Dr Richard Squires. Biological Therapy: What is biological therapy? Biological therapy is the use of a biological compound, not a chemical, to treat disease. It is designed to stimulate or restore the ability of the body’s immune (natural internal defence) system to fight infection and disease. It is also called biological response modifier therapy (BRM), biotherapy or immunotherapy. The therapeutic substances may occur naturally in the body or be made in laboratory cultures rather than chemical synthesis. BRMs are not metabolised like a drug, don’t pass through the liver or kidneys, have a long-lasting action in the body and are very targeted. Which kind of biological therapy and biological response modifiers (BRM) exist? Biological therapy or immunotherapy can be active (where the immune system has to react) or passive (administration of an immunologic molecule like an antibody (Ab) or something similar). -

Clinical Efficacy of Oclacitinib and Lokivetmab in Dogs with Canine Atopic Dermatitis
Original Article pISSN 1598-298X • eISSN 2384-0749 J Vet Clin 2021;38:127-134 https://doi.org/10.17555/jvc.2021.38.3.127 JVCJournal of Veterinary Clinics Clinical Efficacy of Oclacitinib and Lokivetmab in Dogs with Canine Atopic Dermatitis Sora Lee Abstract Canine atopic dermatitis (CAD) is a genetically predisposed inflamma- Taesik Yun tory and pruritic skin disease presenting characteristic clinical features in dogs. Yoonhoi Koo Despite oclacitinib and lokivetmab being commonly used, no study has compared Yeon Chae their efficacies in CAD. This study aimed to compare the efficacy, safety, and con- Dohee Lee trol of CAD-associated pruritus and skin lesions between oclacitinib and lokivet- Dongjoon Choi mab. It also investigated whether switching to lokivetmab from oclacitinib or pred- Yujin Choi nisolone had any benefits. Twenty-five client-owned dogs, newly diagnosed with Hakhyun Kim CAD, were allocated to the oclacitinib (n = 20) and lokivetmab (n = 5) groups and Mhan-Pyo Yang administered oclacitinib (0.4-0.6 mg/kg orally, twice daily for 14 days, then once Byeong-Teck Kang* daily) and lokivetmab (2 mg/kg subcutaneously, every month) for 8 weeks, respec- tively. The switching group included five dogs previously administered with oclac- Laboratory of Veterinary Internal Med- itinib (n = 4) or prednisolone (n = 1) who were switched to lokivetmab directly at icine, College of Veterinary Medicine, the start of the study. The pruritus visual analog scale (PVAS) and Canine Atopic Chungbuk National University, Cheongju 28644, Korea Dermatitis Extent and Severity Index (CADESI-04) values were surveyed at weeks 0, 4, and 8. Oclacitinib and lokivetmab significantly reduced the PVAS and CADE- SI-04 scores. -

Farmaatsia- Terminoloogia Teine, Täiendatud Trükk
Farmaatsia- terminoloogia Teine, täiendatud trükk Graanulid Suspensioon Lahus Emulsioon Pillid Pulber Salv Kreem Aerosool Plaaster Sprei Pastill Tampoon Oblaat Emulsioon Kontsentraat Silmageel Tablett Haavapulk Ninatilgad Kapsel Lakukivi Inhalaator Farmaatsia- terminoloogia Teine, täiendatud trükk Tartu 2019 Koostajad: Toivo Hinrikus, Karin Kogermann, Ott Laius, Signe Leito, Ain Raal, Andres Soosaar, Triin Teppor, Daisy Volmer Keeletoimetaja: Tiina Kuusk Kirjastanud: Ravimiamet Nooruse 1, 50411 Tartu Telefon: +372 737 4140 Faks: +372 737 4142 E-post: [email protected] Esimene trükk 2010 Teine, täiendatud trükk 2019 Raamat on leitav Ravimiameti veebilehelt: www.ravimiamet.ee/farmaatsiaterminoloogia Väljaande refereerimisel või tsiteerimisel palume viidata allikale. ISBN 978-9949-9697-3-9 Sisukord Farmaatsiaterminoloogia Eestis..........................................................................................5 Üldised farmaatsiaalased terminid ...................................................................................10 Euroopa farmakopöa ......................................................................................................... 21 Euroopa farmakopöa sõnastik ..........................................................................................24 Standardterminid ..............................................................................................................29 Ravimvormid .....................................................................................................................29 -

7/23/2021 Human IL-31 Protein, His Tag (MALS Verified) Catalog # IL1-H5247
Human IL-31 Protein, His Tag (MALS verified) Catalog # IL1-H5247 Synonym Formulation IL-31,IL31,Interleukin-31 Lyophilized from 0.22 μm filtered solution in PBS, pH7.4. Normally trehalose is Source added as protectant before lyophilization. Human IL-31, His Tag (IL1-H5247) is expressed from human 293 cells Contact us for customized product form or formulation. (HEK293). It contains AA Ser 24 - Thr 164 (Accession # Q6EBC2-1). Reconstitution Predicted N-terminus: His Molecular Characterization Please see Certificate of Analysis for specific instructions. For best performance, we strongly recommend you to follow the reconstitution protocol provided in the CoA. Storage This protein carries a polyhistidine tag at the N-terminus. For long term storage, the product should be stored at lyophilized state at -20°C The protein has a calculated MW of 17.7 kDa. The protein migrates as 23-33 or lower. kDa under reducing (R) condition (SDS-PAGE) due to glycosylation. Please avoid repeated freeze-thaw cycles. Endotoxin This product is stable after storage at: Less than 1.0 EU per μg by the LAL method. -20°C to -70°C for 12 months in lyophilized state; -70°C for 3 months under sterile conditions after reconstitution. Purity >90% as determined by SDS-PAGE. >90% as determined by SEC-MALS. SDS-PAGE SEC-MALS Human IL-31, His Tag on SDS-PAGE under reducing (R) condition. The gel The purity of Human IL-31, His Tag (Cat. No. IL1-H5247) was more than 90% was stained overnight with Coomassie Blue. The purity of the protein is greater and the molecular weight of this protein is around 15-25 kDa verified by SEC- than 90%.