Np63 Regulates IL-33 and IL-31 Signaling in Atopic Dermatitis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Data Sheet Huil36g (152 A.A.)
Growth Factor Data Sheet GoldBio growth factors are manufactured for RESEARCH USE ONLY and cannot be sold for human consumption! Interleukin-36G (IL36G) is a pro-inflammatory cytokine that plays an important role in the pathophysiology of several diseases. IL36A, IL36B and IL36G (formerly IL1F6, IL1F8, and IL1F9) are IL1 family members that signal through the IL1 receptor family members IL1Rrp2 (IL1RL2) and IL1RAcP. IL36G is secreted when transfected into 293-T cells and could constitute part of an independent signaling system analogous to that of IL1A and IL1B receptor agonist and interleukin-1 receptor type I (IL1R1). Furthermore, IL36G also can function as an agonist of NFκB activation through the orphan IL1- receptor-related protein 2. Human IL36G (152 a.a.) shares 58%, 59%, 68% and 69% amino acid sequence identity with mouse, rat, bovine and equine IL36G, respectively, and 23-57% amino acid sequence identity with other family members. Catalog Number 1110-36F Product Name IL36G (IL-36 gamma), Human (152 a.a.) Recombinant Human Interleukin-36γ IL36G, IL36γ Interleukin 1 Homolog 1 (IL1H1) Interleukin 1-Related Protein 2 (IL1RP2) Interleukin 1 Family, Member 9 (IL1F9) Source Escherichia coli MW ~17.0 kDa (152 amino acids) Sequence SMCKPITGTI NDLNQQVWTL QGQNLVAVPR SDSVTPVTVA VITCKYPEAL EQGRGDPIYL GIQNPEMCLY CEKVGEQPTL QLKEQKIMDL YGQPEPVKPF LFYRAKTGRT STLESVAFPD WFIASSKRDQ PIILTSELGK SYNTAFELNI ND Accession Number Q9NZH8 Purity >95% by SDS-PAGE and HPLC analyses Biological Activity Fully biologically active when compared to standard. The ED50 as determined by its ability to induce IL-8 secretion by human preadipocytes is less than 10 ng/ml, corresponding to a specific activity of >1 × 105 IU/mg. -

Cytokine Nomenclature
RayBiotech, Inc. The protein array pioneer company Cytokine Nomenclature Cytokine Name Official Full Name Genbank Related Names Symbol 4-1BB TNFRSF Tumor necrosis factor NP_001552 CD137, ILA, 4-1BB ligand receptor 9 receptor superfamily .2. member 9 6Ckine CCL21 6-Cysteine Chemokine NM_002989 Small-inducible cytokine A21, Beta chemokine exodus-2, Secondary lymphoid-tissue chemokine, SLC, SCYA21 ACE ACE Angiotensin-converting NP_000780 CD143, DCP, DCP1 enzyme .1. NP_690043 .1. ACE-2 ACE2 Angiotensin-converting NP_068576 ACE-related carboxypeptidase, enzyme 2 .1 Angiotensin-converting enzyme homolog ACTH ACTH Adrenocorticotropic NP_000930 POMC, Pro-opiomelanocortin, hormone .1. Corticotropin-lipotropin, NPP, NP_001030 Melanotropin gamma, Gamma- 333.1 MSH, Potential peptide, Corticotropin, Melanotropin alpha, Alpha-MSH, Corticotropin-like intermediary peptide, CLIP, Lipotropin beta, Beta-LPH, Lipotropin gamma, Gamma-LPH, Melanotropin beta, Beta-MSH, Beta-endorphin, Met-enkephalin ACTHR ACTHR Adrenocorticotropic NP_000520 Melanocortin receptor 2, MC2-R hormone receptor .1 Activin A INHBA Activin A NM_002192 Activin beta-A chain, Erythroid differentiation protein, EDF, INHBA Activin B INHBB Activin B NM_002193 Inhibin beta B chain, Activin beta-B chain Activin C INHBC Activin C NM005538 Inhibin, beta C Activin RIA ACVR1 Activin receptor type-1 NM_001105 Activin receptor type I, ACTR-I, Serine/threonine-protein kinase receptor R1, SKR1, Activin receptor-like kinase 2, ALK-2, TGF-B superfamily receptor type I, TSR-I, ACVRLK2 Activin RIB ACVR1B -

Association of an Interleukin 1B Gene Polymorphism (-511) With
400 LETTER TO JMG Association of an interleukin 1B gene polymorphism (−511) with Parkinson’s disease in Finnish patients K M Mattila, J O Rinne, T Lehtimäki, M Röyttä, J-P Ahonen, M Hurme ............................................................................................................................. J Med Genet 2002;39:400–402 lzheimer’s disease (AD) and Parkinson’s disease (PD) are the two most common neurodegenerative conditions Table 1 Genotype and allele frequencies of the IL1 characterised by the presence of abnormal accumula- gene cluster polymorphisms in AD, PD, and control A patients tion of proteins and loss of neurones in specific regions of the brain. The aetiology and pathogenesis of AD and PD still Controls remain largely unknown. Evidence has emerged that immune Polymorphism AD (n=92) PD (n=52) (n=73) mediated mechanisms may be important in the development − 12 IL1A ( 889) of these disorders, and cytokine interleukin 1 (IL1) is one of *1/*1 42 (0.46) 28 (0.54) 33 (0.45) the mediators suggested to be involved. *1/*2 39 (0.42) 20 (0.38) 29 (0.40) The IL1 family comprises three proteins, the proinflamma- *2/*2 11 (0.12) 4 (0.08) 11 (0.15) tory IL1α and IL1β and their inhibitor IL1 receptor antagonist *1 123 (0.67) 76 (0.73) 95 (0.65) *2 61 (0.32) 28 (0.27) 51 (0.35) (IL1Ra), which are encoded by the IL1A, IL1B, and IL1RN − genes respectively.3 Since IL1 may have a role in AD4–8 and in IL1B ( 511) 910 *1/*1 35 (0.38) 25 (0.48) 24 (0.32) PD, variation in the IL1A, IL1B, and IL1RN genes may be of *1/*2 47 (0.51) 25 (0.48) 30 (0.41) importance in the development of these disorders. -

Genetic Determinants of Circulating Interleukin-1 Receptor Antagonist Levels and Their Association with Glycemic Traits
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Trepo - Institutional Repository of Tampere University GENETIC DETERMINANTS OF CIRCULATING INTERLEUKIN-1 RECEPTOR ANTAGONIST LEVELS AND THEIR ASSOCIATION WITH GLYCEMIC TRAITS Marja-Liisa Nuotio Syventävien opintojen kirjallinen työ Tampereen yliopisto Lääketieteen yksikkö Tammikuu 2015 Tampereen yliopisto Lääketieteen yksikkö NUOTIO MARJA-LIISA: GENETIC DETERMINANTS OF CIRCULATING INTERLEUKIN-1 RECEPTOR ANTAGONIST LEVELS AND THEIR ASSOCIATION WITH GLYCEMIC TRAITS Kirjallinen työ, 57 s. Ohjaaja: professori Mika Kähönen Tammikuu 2015 Avainsanat: sytokiinit, insuliiniresistenssi, tyypin 2 diabetes, tulehdus, glukoosimetabolia, genominlaajuinen assosiaatioanalyysi (GWAS) Tulehdusta välittäviin sytokiineihin kuuluvan interleukiini 1β (IL-1β):n kohonneen systeemisen pitoisuuden on arveltu edesauttavan insuliiniresistenssin kehittymistä ja johtavan haiman β-solujen toimintahäiriöihin. IL-1β:n sisäsyntyisellä vastavaikuttajalla, interleukiini 1 reseptoriantagonistilla (IL-1RA), on puolestaan esitetty olevan suojaava rooli mainittujen fenotyyppien kehittymisessä päinvastaisten vaikutustensa ansiosta. IL-1RA:n suojaavan roolin havainnollistamiseksi työssä Genetic determinants of circulating interleukin-1 receptor antagonist levels and their association with glycemic traits tunnistettiin veren IL-1RA- pitoisuuteen assosioituvia geneettisiä variantteja, minkä jälkeen selvitettiin näiden yhteyttä glukoosi- ja insuliinimetaboliaan liittyvien muuttujien-, sekä -

Comprehensive Association Study of Genetic Variants in the IL-1 Gene Family in Systemic Juvenile Idiopathic Arthritis
Genes and Immunity (2008) 9, 349–357 & 2008 Nature Publishing Group All rights reserved 1466-4879/08 $30.00 www.nature.com/gene ORIGINAL ARTICLE Comprehensive association study of genetic variants in the IL-1 gene family in systemic juvenile idiopathic arthritis CJW Stock1, EM Ogilvie1, JM Samuel1, M Fife1, CM Lewis2 and P Woo1 1Centre for Paediatric and Adolescent Rheumatology, Windeyer Institute for Medical Sciences, University College London, London, UK and 2Guy’s, Kings and St Thomas’ School of Medicine, London, UK Patients with systemic juvenile idiopathic arthritis (sJIA) have a characteristic daily spiking fever and elevated levels of inflammatory cytokines. Members of the interleukin-1 (IL-1) gene family have been implicated in various inflammatory and autoimmune diseases, and treatment with the IL-1 receptor antagonist, Anakinra, shows remarkable improvement in some patients. This work describes the most comprehensive investigation to date of the involvement of the IL-1 gene family in sJIA. A two-stage case–control association study was performed to investigate the two clusters of IL-1 family genes using a tagging single nucleotide polymorphism (SNP) approach. Genotyping data of 130 sJIA patients and 151 controls from stage 1 highlighted eight SNPs in the IL1 ligand cluster region and two SNPs in the IL1 receptor cluster region as showing a significant frequency difference between the populations. These 10 SNPs were typed in an additional 105 sJIA patients and 184 controls in stage 2. Meta-analysis of the genotypes from both stages showed that three IL1 ligand cluster SNPs (rs6712572, rs2071374 and rs1688075) and one IL1 receptor cluster SNP (rs12712122) show evidence of significant association with sJIA. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
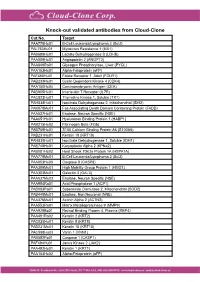
Knock-Out Validated Antibodies from Cloud-Clone Cat.No
Knock-out validated antibodies from Cloud-Clone Cat.No. Target PAA778Hu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAL763Hu01 Myxovirus Resistance 1 (MX1) PAB698Hu01 Lactate Dehydrogenase B (LDHB) PAA009Hu01 Angiopoietin 2 (ANGPT2) PAA849Ra01 Glycogen Phosphorylase, Liver (PYGL) PAA153Hu01 Alpha-Fetoprotein (aFP) PAF460Hu01 Folate Receptor 1, Adult (FOLR1) PAB233Hu01 Cyclin Dependent Kinase 4 (CDK4) PAA150Hu04 Carcinoembryonic Antigen (CEA) PAB905Hu01 Interleukin 7 Receptor (IL7R) PAC823Hu01 Thymidine Kinase 1, Soluble (TK1) PAH838Hu01 Isocitrate Dehydrogenase 2, mitochondrial (IDH2) PAK078Mu01 Fas Associating Death Domain Containing Protein (FADD) PAA537Hu01 Enolase, Neuron Specific (NSE) PAA651Hu01 Hyaluronan Binding Protein 1 (HABP1) PAB215Hu02 Fibrinogen Beta (FGb) PAB769Hu01 S100 Calcium Binding Protein A6 (S100A6) PAB231Hu01 Keratin 18 (KRT18) PAH839Hu01 Isocitrate Dehydrogenase 1, Soluble (IDH1) PAE748Hu01 Karyopherin Alpha 2 (KPNa2) PAB081Hu02 Heat Shock 70kDa Protein 1A (HSPA1A) PAA778Mu01 B-Cell Leukemia/Lymphoma 2 (Bcl2) PAA853Hu03 Caspase 8 (CASP8) PAA399Mu01 High Mobility Group Protein 1 (HMG1) PAA303Mu01 Galectin 3 (GAL3) PAA537Mu02 Enolase, Neuron Specific (NSE) PAA994Ra01 Acid Phosphatase 1 (ACP1) PAB083Ra01 Superoxide Dismutase 2, Mitochondrial (SOD2) PAB449Mu01 Enolase, Non Neuronal (NNE) PAA376Mu01 Actinin Alpha 2 (ACTN2) PAA553Ra01 Matrix Metalloproteinase 9 (MMP9) PAA929Bo01 Retinol Binding Protein 4, Plasma (RBP4) PAA491Ra02 Keratin 2 (KRT2) PAC025Hu01 Keratin 8 (KRT8) PAB231Mu01 Keratin 18 (KRT18) PAC598Hu03 Vanin 1 (VNN1) -

Evolutionary Divergence and Functions of the Human Interleukin (IL) Gene Family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W
UPDATE ON GENE COMPLETIONS AND ANNOTATIONS Evolutionary divergence and functions of the human interleukin (IL) gene family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W. Nebert3* and Vasilis Vasiliou1 1Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences, University of Colorado Denver, Aurora, CO 80045, USA 2Department of Clinical Pharmacy, University of Colorado Denver, Aurora, CO 80045, USA 3Department of Environmental Health and Center for Environmental Genetics (CEG), University of Cincinnati Medical Center, Cincinnati, OH 45267–0056, USA *Correspondence to: Tel: þ1 513 821 4664; Fax: þ1 513 558 0925; E-mail: [email protected]; [email protected] Date received (in revised form): 22nd September 2010 Abstract Cytokines play a very important role in nearly all aspects of inflammation and immunity. The term ‘interleukin’ (IL) has been used to describe a group of cytokines with complex immunomodulatory functions — including cell proliferation, maturation, migration and adhesion. These cytokines also play an important role in immune cell differentiation and activation. Determining the exact function of a particular cytokine is complicated by the influence of the producing cell type, the responding cell type and the phase of the immune response. ILs can also have pro- and anti-inflammatory effects, further complicating their characterisation. These molecules are under constant pressure to evolve due to continual competition between the host’s immune system and infecting organisms; as such, ILs have undergone significant evolution. This has resulted in little amino acid conservation between orthologous proteins, which further complicates the gene family organisation. Within the literature there are a number of overlapping nomenclature and classification systems derived from biological function, receptor-binding properties and originating cell type. -

Interleukin-1And Interleukin-6 Gene Polymorphisms and the Risk of Breast Cancer in Caucasian Women
Human Cancer Biology Interleukin-1 and Interleukin-6 Gene Polymorphisms and the Risk of Breast Cancer in Caucasian Women Lukas A. Hefler,1, 3 Christoph Grimm,1Tilmann Lantzsch,4 Dieter Lampe,4 Sepp Leodolter,1, 3 Heinz Koelbl,5 Georg Heinze,2 Alexander Reinthaller,1Dan Tong-Cacsire,1Clemens Tempfer,1, 3 and Robert Zeillinger1, 3 Abstract Purpose: Genetic polymorphisms of cytokine-encoding genes are known to predispose to malignant disease. Interleukin (IL)-1and IL-6 are crucially involved in breast carcinogenesis. Whether polymorphisms of the genes encoding IL-1 (IL1)andIL-6(IL6) also influence breast cancer risk is unknown. Experimental Design: In the present case-control study, we ascertained three polymorphisms of the IL1 gene cluster [À889 C/T polymorphism of the IL1agene (IL1A), À511C/T polymorphism of the IL1b promoter (IL1B promoter), a polymorphism of IL1b exon 5 (IL1B exon 5 )], an 86-bp repeat in intron 2 of the IL1 receptor antagonist gene (IL1RN), and the À174 G/C polymorphism of the IL6 gene (IL6 ) in 269 patients with breast cancer and 227 healthy controls using PCR and pyrosequencing. Results: Polymorphisms within the IL1 gene cluster and the respective haplotypes were not associated with the presence and the phenotype of breast cancer. The IL6 polymorphism was significantly associated with breast cancer. Odds ratios for women with one or two high-risk alleles versus women homozygous for the low-risk allele were 1.5 (95% confidence interval, 1.04-2.3; P = 0.04) and 2.0 (95% confidence interval, 1.1-3.6; P = 0.02), respectively. -

Interleukin 1-Α: a Modulator of Melanocyte Homeostasis in Vitiligo
nalytic A al & B y i tr o Singh et al., Biochem Anal Biochem 2016, 5:2 s c i h e m m Biochemistry & e DOI: 10.4172/2161-1009.1000273 i h s c t r o i y B ISSN: 2161-1009 Analytical Biochemistry Research Article Open Access Interleukin 1-α: A Modulator of Melanocyte Homeostasis in Vitiligo Mala Singh, Mohmmad Shoab Mansuri, Mansi A Parasrampuria and Rasheedunnisa Begum* Department of Biochemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara, Gujarat -390002, India Abstract Background: Vitiligo is a hypomelanotic autoimmune skin disorder arising from a breakdown in immunological self-tolerance, which leads to aberrant immune responses against melanocytes leading to selective destruction of melanocytes. High levels of Interleukin 1(IL1) have been reported in various autoimmune disorders including vitiligo. The aim of present study was to explore the effect of IL1-α on melanocyte biology by monitoring the melanocyte viability, IL1R1 membrane expression, melanogenesis (TYR, TYRP1 and MITF-M) and other immuno modulatory molecules (IL1A, IL1B, IL1RN,IL1R1 IL8, TNFA, IL6, and ICAM1) upon exogenous stimulation of IL1- α on primary cultured normal human melanocytes (NHM). Materials and methods: Melanocytes were isolated from normal human skin biopsies and cultured in vitro. The NHMs were treated with IL1-α (0-100 ng/ml) and cell viability was monitored by MTT assay, IL1R1 membrane expression was monitored using flow cytometry and relative gene expression was performed using semi-quantitative RT-PCR. Results: The dose dependent effect of IL1-α on melanocytes showed ~12% melanocyte death and ~22% increase in IL1R1 membrane expression upon 100 ng/ml IL1-α treatment for 48 hrs. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

7/23/2021 Human IL-31 Protein, His Tag (MALS Verified) Catalog # IL1-H5247
Human IL-31 Protein, His Tag (MALS verified) Catalog # IL1-H5247 Synonym Formulation IL-31,IL31,Interleukin-31 Lyophilized from 0.22 μm filtered solution in PBS, pH7.4. Normally trehalose is Source added as protectant before lyophilization. Human IL-31, His Tag (IL1-H5247) is expressed from human 293 cells Contact us for customized product form or formulation. (HEK293). It contains AA Ser 24 - Thr 164 (Accession # Q6EBC2-1). Reconstitution Predicted N-terminus: His Molecular Characterization Please see Certificate of Analysis for specific instructions. For best performance, we strongly recommend you to follow the reconstitution protocol provided in the CoA. Storage This protein carries a polyhistidine tag at the N-terminus. For long term storage, the product should be stored at lyophilized state at -20°C The protein has a calculated MW of 17.7 kDa. The protein migrates as 23-33 or lower. kDa under reducing (R) condition (SDS-PAGE) due to glycosylation. Please avoid repeated freeze-thaw cycles. Endotoxin This product is stable after storage at: Less than 1.0 EU per μg by the LAL method. -20°C to -70°C for 12 months in lyophilized state; -70°C for 3 months under sterile conditions after reconstitution. Purity >90% as determined by SDS-PAGE. >90% as determined by SEC-MALS. SDS-PAGE SEC-MALS Human IL-31, His Tag on SDS-PAGE under reducing (R) condition. The gel The purity of Human IL-31, His Tag (Cat. No. IL1-H5247) was more than 90% was stained overnight with Coomassie Blue. The purity of the protein is greater and the molecular weight of this protein is around 15-25 kDa verified by SEC- than 90%.