Engineering and Delivery of Synthetic Chromatin Effectors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
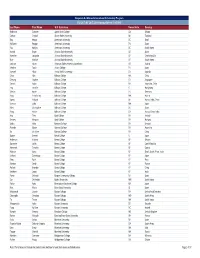
Gilman Fall 2017 Recipients for Website (1.4.18).Xlsx
Benjamin A. Gilman International Scholarship Program Fall 2017/AY 2017-2018 Awards Offered (1/4/2018) Last Name First Name U.S. Institution Home State Country Robinson Summer Agnes Scott College GA Ghana Cotton Crystal Alcorn State University MI Thailand Roy Kenya American University DC Brazil Williams Reagan American University TN Spain You Kaitlyn American University DC South Korea Arriola Bryan Arizona State University AZ Spain Kaercher Jacquelin Arizona State University AZ Czech Republic Ruiz Maribel Arizona State University AZ South Korea Jackson Julian Arkansas State University, Jonesboro AR Austria Medina Paola Austin College TX Spain Samuel Alicia Azusa Pacific University CA Uganda Chau Alex Babson College MA China Cheung Daphne Babson College CA Singapore Dennis Aidan Babson College NY Argentina, Chile Eng Jennifer Babson College CT Hong Kong Macias Karen Babson College TX Germany Ning Priscilla Joy Babson College MA Austria Spears Mikayla Babson College WI Russia, India, China Stetson Lydia Babson College MA Japan West Christopher Babson College LA Spain Yang Austin Babson College CA Russia, China, India Hoy Theo Bard College NY Iceland Kenney Meagan Bard College NY Hungary Gadio Haby Barnard College NY Senegal Paredes Elanie Barnard College NY Argentina Xu Christine Barnard College NY China Baxter Everett Beloit College IL Japan Anderson Kristina Berea College KY Bhutan Bannister Caleb Berea College KY Czech Republic Hammett Timothy Berea College KY Cyprus Hobson Kyree Berea College KY Brazil, South Africa, India Jenkins Dominique -

Crime, Law Enforcement, and Punishment
Shirley Papers 48 Research Materials, Crime Series Inventory Box Folder Folder Title Research Materials Crime, Law Enforcement, and Punishment Capital Punishment 152 1 Newspaper clippings, 1951-1988 2 Newspaper clippings, 1891-1938 3 Newspaper clippings, 1990-1993 4 Newspaper clippings, 1994 5 Newspaper clippings, 1995 6 Newspaper clippings, 1996 7 Newspaper clippings, 1997 153 1 Newspaper clippings, 1998 2 Newspaper clippings, 1999 3 Newspaper clippings, 2000 4 Newspaper clippings, 2001-2002 Crime Cases Arizona 154 1 Cochise County 2 Coconino County 3 Gila County 4 Graham County 5-7 Maricopa County 8 Mohave County 9 Navajo County 10 Pima County 11 Pinal County 12 Santa Cruz County 13 Yavapai County 14 Yuma County Arkansas 155 1 Arkansas County 2 Ashley County 3 Baxter County 4 Benton County 5 Boone County 6 Calhoun County 7 Carroll County 8 Clark County 9 Clay County 10 Cleveland County 11 Columbia County 12 Conway County 13 Craighead County 14 Crawford County 15 Crittendon County 16 Cross County 17 Dallas County 18 Faulkner County 19 Franklin County Shirley Papers 49 Research Materials, Crime Series Inventory Box Folder Folder Title 20 Fulton County 21 Garland County 22 Grant County 23 Greene County 24 Hot Springs County 25 Howard County 26 Independence County 27 Izard County 28 Jackson County 29 Jefferson County 30 Johnson County 31 Lafayette County 32 Lincoln County 33 Little River County 34 Logan County 35 Lonoke County 36 Madison County 37 Marion County 156 1 Miller County 2 Mississippi County 3 Monroe County 4 Montgomery County -

Helsinki in Early Twentieth-Century Literature Urban Experiences in Finnish Prose Fiction 1890–1940
lieven ameel Helsinki in Early Twentieth-Century Literature Urban Experiences in Finnish Prose Fiction 1890–1940 Studia Fennica Litteraria The Finnish Literature Society (SKS) was founded in 1831 and has, from the very beginning, engaged in publishing operations. It nowadays publishes literature in the fields of ethnology and folkloristics, linguistics, literary research and cultural history. The first volume of the Studia Fennica series appeared in 1933. Since 1992, the series has been divided into three thematic subseries: Ethnologica, Folkloristica and Linguistica. Two additional subseries were formed in 2002, Historica and Litteraria. The subseries Anthropologica was formed in 2007. In addition to its publishing activities, the Finnish Literature Society maintains research activities and infrastructures, an archive containing folklore and literary collections, a research library and promotes Finnish literature abroad. Studia fennica editorial board Pasi Ihalainen, Professor, University of Jyväskylä, Finland Timo Kaartinen, Title of Docent, Lecturer, University of Helsinki, Finland Taru Nordlund, Title of Docent, Lecturer, University of Helsinki, Finland Riikka Rossi, Title of Docent, Researcher, University of Helsinki, Finland Katriina Siivonen, Substitute Professor, University of Helsinki, Finland Lotte Tarkka, Professor, University of Helsinki, Finland Tuomas M. S. Lehtonen, Secretary General, Dr. Phil., Finnish Literature Society, Finland Tero Norkola, Publishing Director, Finnish Literature Society Maija Hakala, Secretary of the Board, Finnish Literature Society, Finland Editorial Office SKS P.O. Box 259 FI-00171 Helsinki www.finlit.fi Lieven Ameel Helsinki in Early Twentieth- Century Literature Urban Experiences in Finnish Prose Fiction 1890–1940 Finnish Literature Society · SKS · Helsinki Studia Fennica Litteraria 8 The publication has undergone a peer review. The open access publication of this volume has received part funding via a Jane and Aatos Erkko Foundation grant. -

Appendix 1. Categorization of Cigarette Brands As Either Premium Or Discount
Appendix 1. Categorization of Cigarette Brands as either Premium or Discount Category Name of Cigarette Brand Premium Accord, American Spirit, Barclay, Belair, Benson & Hedges, Camel, Capri, Carlton, Chesterfield, Davidoff, Du Maurier, Dunhill, Dunhill International, Eve, Kent, Kool, L&M, Lark, Lucky Strike, Marlboro, Max, Merit, Mild Seven, More, Nat Sherman, Newport, Now, Parliament, Players, Quest, Rothman’s, Salem, Sampoerna, Saratoga, Tareyton, True, Vantage, Virginia Slims, Winston, Raleigh, Business Club Full Flavor, Ronhill, Dreams Discount 24/7, 305, 1839, A1, Ace, Allstar, Allway Save, Alpine, American, American Diamond, American Hero, American Liberty, Arrow, Austin, Axis, Baileys, Bargain Buy, Baron, Basic, Beacon, Berkeley, Best Value, Black Hawk, Bonus Value, Boston, Bracar, Brand X, Brave, Brentwood, Bridgeport, Bronco, Bronson, Bucks, Buffalo, BV, Calon, Cambridge, Campton, Cannon, Cardinal, Carnival, Cavalier, Champion, Charter, Checkers, Cherokee, Cheyenne, Cimarron, Circle Z, Class A, Classic, Cobra, Complete, Corona, Courier, CT, Decade, Desert Gold, Desert Sun, Discount, Doral, Double Diamond, DTC, Durant, Eagle, Echo, Edgefield, Epic, Esquire, Euro, Exact, Exeter, First Choice, First Class, Focus, Fortuna, Galaxy Pro, Gauloises, Generals, Generic/Private Label, Geronimo, Gold Coast, Gold Crest, Golden Bay, Golden, Golden Beach, Golden Palace, GP, GPC, Grand, Grand Prix, G Smoke, GT Ones, Hava Club, HB, Heron, Highway, Hi-Val, Jacks, Jade, Kentucky Best, King Mountain, Kingsley, Kingston, Kingsport, Knife, Knights, -

INTERNATIONAL CIGARETTE PACKAGING STUDY Summary
INTERNATIONAL CIGARETTE PACKAGING STUDY Summary Technical Report June 2013 TABLE OF CONTENTS RESEARCH TEAM ................................................................................................................... iv 1.0 INTRODUCTION ............................................................................................................... 1 2.0 STUDY PROTOCOL ........................................................................................................... 1 2.1 OVERVIEW ............................................................................................................ 1 2.2 SAMPLE AND RECRUITMENT ................................................................................. 2 3.0 STUDY CONTENT ............................................................................................................. 3 3.1 STUDY 1: HEALTH WARNING MESSAGES ............................................................... 3 3.2 STUDY 2: CIGARETTE PACKAGING ......................................................................... 4 4.0 MEASURES...................................................................................................................... 6 4.1 QUESTIONNAIRE DEVELOPMENT .......................................................................... 6 4.2 QUESTIONNAIRE CONTENT ................................................................................... 6 5.0 SAMPLE INFORMATION ................................................................................................... 9 REFERENCES ........................................................................................................................ -

A Rhetorical Analysis of Selected Sermons by Sam Jones During His Emergence As a National Figure, 1872-1885
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1980 A Rhetorical Analysis of Selected Sermons by Sam Jones During His Emergence as a National Figure, 1872-1885. Herman Daniel Champion Jr Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Champion, Herman Daniel Jr, "A Rhetorical Analysis of Selected Sermons by Sam Jones During His Emergence as a National Figure, 1872-1885." (1980). LSU Historical Dissertations and Theses. 3553. https://digitalcommons.lsu.edu/gradschool_disstheses/3553 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This was produced from a copy of a document sent to us for microfilming. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help you understand markings or notations which may appear on this reproduction. 1. The sign or “target” for pages apparently lacking from the document photographed is “Missing Page(s)”. If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure you of complete continuity. 2. -

Associate Leadership Institute
diversity Associate Leadership Institute 2018 PARTICIPANT DIRECTORY www.nycbar.org/ALI 2018 FELLOWS INDEX Adam Acosta, White & Case LLP Rashida Adams, White & Case LLP Dupe Adegoke, Reed Smith LLP Randa Adra, Crowell & Moring LLP Nelly Almeida, Milbank, Tweed, Hadley & McCloy LLP Sarah Anstey, Fragomen, Del Rey, Bernsen & Loewy, LLP Christopher Avellaneda, Schulte Roth & Zabel LLP Nairuby Beckles, Paul, Weiss, Rifkind, Wharton & Garrison LLP Camille Bent, BakerHostetler LLP Sally Bergmann, Debevoise & Plimpton LLP Tsedey Bogale, Reed Smith LLP Adrienne Bradley, Sullivan & Cromwell LLP Amanda A. Butler-Jones, Akin Gump Strauss Hauer & Feld LLP Marcie Cleary, Frankfurt Kurnit Klein & Selz PC Marissa Comart, Davis & Gilbert LLP Elizabeth Dahill, Seyfarth Shaw LLP Christopher Davis, Kramer Levin Naftalis & Frankel LLP Ekta Dharia, MoloLamken LLP Audra Dowless, Schulte Roth & Zabel LLP Aleesha Fowler, McGuireWoods LLP Luke Frankson, Sidley Austin LLP Rebecca R. Friedman, Kasowitz Benson Torres LLP Sharonmoyee Goswami, Cravath, Swaine & Moore LLP Chas Hamilton, Paul, Weiss, Rifkind, Wharton & Garrison LLP Brian Harris, Ropes & Gray LLP Antonio Haynes, Davis Polk & Wardwell LLP Simone Hicks, Debevoise & Plimpton LLP Susan Hu, Arnold & Porter LLP Keith A. James Jr., Paul, Weiss, Rifkind, Wharton & Garrison LLP Aileen Kim, Weil, Gotshal & Manges LLP Christina Kim, Davis Wright Tremaine LLP 1 | City Bar Associate Leadership Institute | 2018 Julia Kim, Sullivan & Cromwell LLP Joyce Kwok, Sullivan & Cromwell LLP Cassandra Labbees, Epstein Becker and Green PC Justin Lee, White & Case LLP Victoria Lee, WilmerHale Gary Lo, Davis Polk & Wardwell LLP Deborah Kemi Martin, Dechert LLP Katie McShane, Cadwalader Wickersham & Taft LLP Silvia Medina, White & Case LLP Marco Molina, BakerHostetler LLP Nadine F. Mompremier, Ropes & Gray LLP Rohit Nafday, Wachtell, Lipton, Rosen and Katz Marianna Ofosu, Wachtell, Lipton, Rosen and Katz Byron Pacheco, Boies Schiller Flexner LLP Andrew P. -

Monitoring Changing Tobacco Use Behaviors: 2000 - 2014
Monitoring Changing Tobacco Use Behaviors: 2000 - 2014 Maryland Department of Health and Mental Hygiene Cigarette Restitution Fund Center for Tobacco Prevention and Control State Fiscal Year 2015 Larry Hogan Governor State of Maryland Boyd Rutherford Lieutenant Governor State of Maryland Van Mitchell Secretary Department of Health and Mental Hygiene Statutory Authority and Requirements Maryland’s Health-General Article, Title 13, Subtitle 10, requires the Maryland Department of Health and Mental Hygiene (DHMH) to conduct a biennial tobacco study and report specific findings to the Maryland Governor and the General Assembly. The appendices to this report provide the detailed data for indicators DHMH is required to report in its biennial tobacco study for underage youth. THIS PAGE HAS BEEN LEFT BLANK INTENTIONALLY 1 Table of Contents Suggested Citation ......................................................................................................................... 5 Cover Letter ..................................................................................................................................... 6 In Brief ............................................................................................................................................... 8 Commonly Used Acronyms Found in this Report ..................................................................... 11 About this Report .......................................................................................................................... 12 Data in this -

Unobtrusive Observations of Cigarette Smoking
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 6-1984 Unobtrusive Observations of Cigarette Smoking Robert Fisher Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Recommended Citation Fisher, Robert, "Unobtrusive Observations of Cigarette Smoking. " PhD diss., University of Tennessee, 1984. https://trace.tennessee.edu/utk_graddiss/5326 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Robert Fisher entitled "Unobtrusive Observations of Cigarette Smoking." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Psychology. William S. Verplanck, Major Professor We have read this dissertation and recommend its acceptance: Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) To the Graduate Council: I am submitting herewith a dissertation written by Robert Fisher entitled "Unobtrusive Observations of Cigarette Smoking." I have exam ined the final copy of this dissertation for form and content and rec ommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Psychology. -

TOBACCO (To Be Filed by Manufacturers Who Do Not Participate in the Master Settlement Agreement) for Sales Made Within Michigan During the 2002 Calendar Year
ANNUAL CERTIFICATION OF COMPLIANCE FOR MANUFACTURER OF CIGARETTES AND/OR “ROLL-YOUR-OWN” TOBACCO (To be filed by manufacturers who do not participate in the Master Settlement Agreement) For sales made within Michigan during the 2002 calendar year Amendments in 2002 to the Tobacco Products Tax Act (1993 PA 327) support enforcement of the escrow requirements for tobacco product manufacturers who are not participating in the tobacco Master Settlement Agreement (1999 PA 244). The amendments require that non-participating manufacturers (NPM’s) provide to the State, and anyone who sells their cigarettes and/or ‘roll-your-own’ tobacco for consumption in Michigan, an annual certification of compliance. By providing the certification, they are attesting to the fact that they have met their escrow obligation under Act 244. The amendments prohibit anyone in Michigan from acquiring, possessing, or selling cigarettes and/or ‘roll-your-own’ tobacco manufactured by an NPM who has failed to provide the certification. The Act provides severe penalties for NPM’s, or anyone selling the cigarettes and/or ‘roll-your-own’ tobacco of NPM’s, for failure to comply with the requirements. The cigarettes, including ‘roll-your-own’ tobacco, of an NPM who has failed to provide the annual certification required by the Tobacco Products Tax Act are subject to seizure or confiscation from anyone in possession of them. If an NPM or any other person does not comply with these requirements, they may be subject to a civil fine not to exceed $1,000.00 per violation, in addition to other penalties that may be imposed under this act or the revenue act. -

Information to Users
INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. Each original is also photographed in one exposure and is included in reduced form at the back of the book. Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6" x 9" black and white photographic prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order. UMI University Microfilms International A Bell & Howell Information Company 300 Nortfi Zeeb Road, Ann Arbor, Ml 48106-1346 USA 313/761-4700 800/521-0600 Order Number 9227230 The development and performance of chromium/ reactive element-modified aluminide diffusion coatings by chloride-activated pack cementation Bianco, Robert, Ph.D. -

Emerging Research on Potential Health Effects of Electronic Cigarettes Secondhand Exposure Flavorings
Emerging Research on Potential Health Effects of Electronic Cigarettes The proposed Orange County Board of Health rule to prohibit use in enclosed areas of restaurants and bars is primarily concerned with preventing secondhand exposure and not with the role of e-cigarettes on smoking promotion vs. cessation. Therefore, the research cited below focuses on the chemical composition of e-liquids and aerosol, toxicology studies, the potential for secondhand exposure, and documented health effects. There are additional bodies of research on the effectiveness and safety of e-cigarettes for smoking cessation that are not listed here. The emerging body of research on e-cigarettes suggests that emitted aerosol may contain potentially harmful chemicals in addition to nicotine or other drugs. Secondhand Exposure Research demonstrates the potential for secondhand exposure to e-cigarette aerosol through biomarkers of nicotine exposure, as well as through studies done in controlled indoor conditions. Ballbé M, Martínez-Sánchez JM. (2014). Cigarettes vs. E-Cigarettes: Passive Exposure at Home Measured by Means of Airborne Marker and Biomarkers. Environmental Research. 135:76–80. This study showed that non-smokers passively exposed to e-cigarettes absorb nicotine. This study characterized passive exposure to nicotine from e-cigarette vapor and conventional cigarette smoke at home among non-smokers under real-use conditions. The airborne markers were statistically higher in conventional cigarette homes than in e-cigarettes homes (5.7 times higher). However, concentrations of both biomarkers among non- smokers exposed to conventional cigarette smoke and e-cigarette vapor were statistically similar (only 2 and 1.4 times higher, respectively).The levels of airborne nicotine and cotinine concentrations in the homes with e-cigarette users were statistically higher than control homes.