Information to Users
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
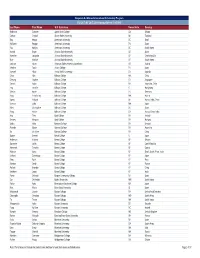
Gilman Fall 2017 Recipients for Website (1.4.18).Xlsx
Benjamin A. Gilman International Scholarship Program Fall 2017/AY 2017-2018 Awards Offered (1/4/2018) Last Name First Name U.S. Institution Home State Country Robinson Summer Agnes Scott College GA Ghana Cotton Crystal Alcorn State University MI Thailand Roy Kenya American University DC Brazil Williams Reagan American University TN Spain You Kaitlyn American University DC South Korea Arriola Bryan Arizona State University AZ Spain Kaercher Jacquelin Arizona State University AZ Czech Republic Ruiz Maribel Arizona State University AZ South Korea Jackson Julian Arkansas State University, Jonesboro AR Austria Medina Paola Austin College TX Spain Samuel Alicia Azusa Pacific University CA Uganda Chau Alex Babson College MA China Cheung Daphne Babson College CA Singapore Dennis Aidan Babson College NY Argentina, Chile Eng Jennifer Babson College CT Hong Kong Macias Karen Babson College TX Germany Ning Priscilla Joy Babson College MA Austria Spears Mikayla Babson College WI Russia, India, China Stetson Lydia Babson College MA Japan West Christopher Babson College LA Spain Yang Austin Babson College CA Russia, China, India Hoy Theo Bard College NY Iceland Kenney Meagan Bard College NY Hungary Gadio Haby Barnard College NY Senegal Paredes Elanie Barnard College NY Argentina Xu Christine Barnard College NY China Baxter Everett Beloit College IL Japan Anderson Kristina Berea College KY Bhutan Bannister Caleb Berea College KY Czech Republic Hammett Timothy Berea College KY Cyprus Hobson Kyree Berea College KY Brazil, South Africa, India Jenkins Dominique -

Associate Leadership Institute
diversity Associate Leadership Institute 2018 PARTICIPANT DIRECTORY www.nycbar.org/ALI 2018 FELLOWS INDEX Adam Acosta, White & Case LLP Rashida Adams, White & Case LLP Dupe Adegoke, Reed Smith LLP Randa Adra, Crowell & Moring LLP Nelly Almeida, Milbank, Tweed, Hadley & McCloy LLP Sarah Anstey, Fragomen, Del Rey, Bernsen & Loewy, LLP Christopher Avellaneda, Schulte Roth & Zabel LLP Nairuby Beckles, Paul, Weiss, Rifkind, Wharton & Garrison LLP Camille Bent, BakerHostetler LLP Sally Bergmann, Debevoise & Plimpton LLP Tsedey Bogale, Reed Smith LLP Adrienne Bradley, Sullivan & Cromwell LLP Amanda A. Butler-Jones, Akin Gump Strauss Hauer & Feld LLP Marcie Cleary, Frankfurt Kurnit Klein & Selz PC Marissa Comart, Davis & Gilbert LLP Elizabeth Dahill, Seyfarth Shaw LLP Christopher Davis, Kramer Levin Naftalis & Frankel LLP Ekta Dharia, MoloLamken LLP Audra Dowless, Schulte Roth & Zabel LLP Aleesha Fowler, McGuireWoods LLP Luke Frankson, Sidley Austin LLP Rebecca R. Friedman, Kasowitz Benson Torres LLP Sharonmoyee Goswami, Cravath, Swaine & Moore LLP Chas Hamilton, Paul, Weiss, Rifkind, Wharton & Garrison LLP Brian Harris, Ropes & Gray LLP Antonio Haynes, Davis Polk & Wardwell LLP Simone Hicks, Debevoise & Plimpton LLP Susan Hu, Arnold & Porter LLP Keith A. James Jr., Paul, Weiss, Rifkind, Wharton & Garrison LLP Aileen Kim, Weil, Gotshal & Manges LLP Christina Kim, Davis Wright Tremaine LLP 1 | City Bar Associate Leadership Institute | 2018 Julia Kim, Sullivan & Cromwell LLP Joyce Kwok, Sullivan & Cromwell LLP Cassandra Labbees, Epstein Becker and Green PC Justin Lee, White & Case LLP Victoria Lee, WilmerHale Gary Lo, Davis Polk & Wardwell LLP Deborah Kemi Martin, Dechert LLP Katie McShane, Cadwalader Wickersham & Taft LLP Silvia Medina, White & Case LLP Marco Molina, BakerHostetler LLP Nadine F. Mompremier, Ropes & Gray LLP Rohit Nafday, Wachtell, Lipton, Rosen and Katz Marianna Ofosu, Wachtell, Lipton, Rosen and Katz Byron Pacheco, Boies Schiller Flexner LLP Andrew P. -

Monitoring Changing Tobacco Use Behaviors: 2000 - 2014
Monitoring Changing Tobacco Use Behaviors: 2000 - 2014 Maryland Department of Health and Mental Hygiene Cigarette Restitution Fund Center for Tobacco Prevention and Control State Fiscal Year 2015 Larry Hogan Governor State of Maryland Boyd Rutherford Lieutenant Governor State of Maryland Van Mitchell Secretary Department of Health and Mental Hygiene Statutory Authority and Requirements Maryland’s Health-General Article, Title 13, Subtitle 10, requires the Maryland Department of Health and Mental Hygiene (DHMH) to conduct a biennial tobacco study and report specific findings to the Maryland Governor and the General Assembly. The appendices to this report provide the detailed data for indicators DHMH is required to report in its biennial tobacco study for underage youth. THIS PAGE HAS BEEN LEFT BLANK INTENTIONALLY 1 Table of Contents Suggested Citation ......................................................................................................................... 5 Cover Letter ..................................................................................................................................... 6 In Brief ............................................................................................................................................... 8 Commonly Used Acronyms Found in this Report ..................................................................... 11 About this Report .......................................................................................................................... 12 Data in this -

Emerging Research on Potential Health Effects of Electronic Cigarettes Secondhand Exposure Flavorings
Emerging Research on Potential Health Effects of Electronic Cigarettes The proposed Orange County Board of Health rule to prohibit use in enclosed areas of restaurants and bars is primarily concerned with preventing secondhand exposure and not with the role of e-cigarettes on smoking promotion vs. cessation. Therefore, the research cited below focuses on the chemical composition of e-liquids and aerosol, toxicology studies, the potential for secondhand exposure, and documented health effects. There are additional bodies of research on the effectiveness and safety of e-cigarettes for smoking cessation that are not listed here. The emerging body of research on e-cigarettes suggests that emitted aerosol may contain potentially harmful chemicals in addition to nicotine or other drugs. Secondhand Exposure Research demonstrates the potential for secondhand exposure to e-cigarette aerosol through biomarkers of nicotine exposure, as well as through studies done in controlled indoor conditions. Ballbé M, Martínez-Sánchez JM. (2014). Cigarettes vs. E-Cigarettes: Passive Exposure at Home Measured by Means of Airborne Marker and Biomarkers. Environmental Research. 135:76–80. This study showed that non-smokers passively exposed to e-cigarettes absorb nicotine. This study characterized passive exposure to nicotine from e-cigarette vapor and conventional cigarette smoke at home among non-smokers under real-use conditions. The airborne markers were statistically higher in conventional cigarette homes than in e-cigarettes homes (5.7 times higher). However, concentrations of both biomarkers among non- smokers exposed to conventional cigarette smoke and e-cigarette vapor were statistically similar (only 2 and 1.4 times higher, respectively).The levels of airborne nicotine and cotinine concentrations in the homes with e-cigarette users were statistically higher than control homes. -

Vaping and E-Cigarettes: Adding Fuel to the Coronavirus Fire?
Vaping and e-cigarettes: Adding fuel to the coronavirus fire? abcnews.go.com/Health/vaping-cigarettes-adding-fuel-coronavirus-fire/story By Dr. Chloë E. Nunneley 26 March 2020, 17:04 6 min read Vaping and e-cigarettes: Adding fuel to the coronavirus fire?Because vaping can cause dangerous lung and respiratory problems, experts say it makes sense that the habit could aggravate the symptoms of COVID-19. New data released by the Centers for Disease Control and Prevention last week warns that young people may be more impacted by COVID-19 than was initially thought, with patients under the age of 45 comprising more than a third of all cases, and one in five of those patients requiring hospitalization. Although scientists still don’t have good data to explain exactly why some young people are getting very sick from the novel coronavirus, some experts are now saying that the popularity of e-cigarettes and vaping could be making a bad situation even worse. Approximately one in four teens in the United States vapes or smokes e-cigarettes, with the FDA declaring the teenage use of these products a nationwide epidemic and the CDC warning about a life-threatening vaping illness called EVALI, or “E-cigarette or Vaping- Associated Lung Injury.” Public health experts believe that conventional cigarette smokers are likely to have more serious illness if they become infected with COVID-19, according to the World Health Organization. Because vaping can also cause dangerous lung and respiratory problems, 1/4 experts say it makes sense that the habit could aggravate the symptoms of COVID-19, although they will need longer-term studies to know for sure. -

Louisville, Coal Creek, Lafayette Update, and Superior, Boulder Colorado, County Cemeteries
Louisville, Coal Creek, Lafayette Update, and Superior, Boulder Colorado, County Cemeteries Colorado State Society National Society Daughters of the American Revolution Genealogical Records Committee Report: Series 1. Volume 2 Louisville Cemetery, Coal Creek Cemetery, and Superior Cemetery Headstone Inscriptions Indian Peaks Chapter, NSDAR Karen Yudnich, Chapter GRC Chair Tyler Hancock, Colorado State GRC Chair Jeannine Dobbins, Colorado State Regent Ann Turner Dillon, President General, National Society Daughters of the American Revolution Sue Tener Thompson, National Chair Genealogical Records Committee 2018 ©2018 National Society Daughters of the American Revolution. No portion of this book may be copied in any form, electronically, photographically, digitally, or otherwise, without the express written permission of the Genealogical Records Committee, DAR Library, National Society Daughters of the American Revolution 1776 D Street NW, Washington, DC 2006-5303. Louisville, Coal Creek, Lafayette and Superior Cemeteries 1820-2018 Compiled by: Members of the Indian Peaks Chapter NSDAR Melinda Medrick-Nye Karen Marrill Kathy MacQueen Terri Stone Gayle Asbury Kristen Bardsley Pat Winters Rani Machoi Carol Marsh Cherry Moore Bob Yudnich Tambi Gilmore Allie Golon Patty Rocha I dedicate this book to my mother, Dorothy Negley, the oldest organizing member of Indian Peaks Chapter. Karen Yudnich Indian Peaks Chapter NSDAR Genealogical Records Committee Chair 2 Table of Contents Louisville Cemetery ....................................................................................................................... -

Why Do International Students Avoid Communicating with Americans?
Journal of International Students, 7(3) 2017 Peer-Reviewed Article ISSN: 2162-3104 Print/ ISSN: 2166-3750 Online Volume 7, Issue 3 (2017), pp. 555-582 © Journal of International Students http://jistudents.org/ doi:10.5281/zenodo.570023 Why Do International Students Avoid Communicating with Americans? I-Ching Wang Teachers College, Columbia University, USA Janet N. Ahn William Paterson University, USA Hyojin J. Kim Teachers College, Columbia University, USA Xiaodong Lin-Siegler Teachers College, Columbia University, USA ABSTRACT We explore how the communication concerns of non-native English speakers (NNS) and Americans relate to their perceptions of each other and decisions to interact. NNS identified their concerns in communicating with Americans, the perceived causes of their concerns, and the strategies they would employ to address these concerns. Americans noted their perceptions of NNS’ (heavy and mild) accents and identified factors that influence their perceptions of NNS and decisions to communicate with NNS. Results revealed that the more NNS attribute their communication concerns to Americans’ bias, the more likely they would avoid interacting with Americans. Results also suggested that Americans show a more favorable attitude toward mild accent NNS. We discuss the implications these findings have for international education and future research. Keywords: communication, ESL, foreign accent, intercultural competence, perceived bias Over the past decade, the number of international students enrolling in higher education in America has been growing rapidly (Institute of International Education, 2015). Approximately 5 million international students are enrolled in American colleges and universities (OECD, 2015) and the number is projected to reach 8-10 million by the year 2020 (Forest, 2007; Phakiti, Hirsh & Woodrow, 2013). -

The Tobacco Industry in NZ
The Tobacco Industry in New Zealand: A Case Study of the Behaviour of Multinational Companies George Thomson Dr Nick Wilson Public Health Monograph Series No. 6 Department of Public Health Wellington School of Medicine and Health Sciences February 2002 2 Copyright All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording, or otherwise without the prior permission of the authors Public Health Monograph Series No. 6 ISSN 1175 - 7817 ISBN 0 - 473 - 08415 - 5 (Paper version) ISBN 0 - 473 - 08414 - 7 (Electronic version) Published by the Department of Public Health Wellington School of Medicine and Health Sciences PO Box 7343 Wellington South Wellington New Zealand Electronic version at: http://www.wnmeds.ac.nz/academic/dph/docs.htm “If you want to do something about malaria, you have to study mosquitoes. And if you want to do something about lung cancer, you have to study the tobacco industry.” Professor Stan Glantz. In: Noah T. Funding of study of tobacco firms is periled in House. The Wall Street Journal 7 August 1995. 3 The tobacco industry and its allies said it: “... nicotine is addictive. We are, then, in the business of selling nicotine, ….” (Addison Yeaman, Brown & Williamson vice president and general counsel, 1963. Cited in Lewan 1998) “Today’s teenager is tomorrow’s potential regular customer, and the overwhelming majority of smokers first begin to smoke while in their teens … The smoking patterns of teenagers are particularly important to Philip Morris ...” (1981 report sent from researcher Myron E Johnston to Robert B Seligman, then vice president of research and development at Philip Morris in Richmond, Virginia. -

Doğal Tekellerde Özelleştirme Ve Regülasyon
DPT – UZMANLIK TEZLERİ DOĞAL TEKELLERDE ÖZELLEŞTİRME VE REGÜLASYON TEMMUZ 1996 YAYIN NO : DPT: 2455 DOĞAL TEKELLERDE ÖZELLEŞTİRME VE REGÜLASYON Recep ÇAKAL Uzmanlõk Tezi İKTİSADİ SEKTÖRLER VE KOORDİNASYON GENEL MÜDÜRLÜĞÜ SEKTÖR PROGRAMLARI DAİRESİ TEMMUZ 1996 Çakal Doğal Tekellerde Özelleştirme ve Regülasyon ISBN 975 – 19 – 1447-7 (basõlõ nüsha) Bu Çalõşma Devlet Planlama Teşkilatõnõn görüşlerini yansõtmaz. Sorumluluğu yazarõna aittir. Yayõn ve referans olarak kullanõlmasõ Devlet Planlama Teşkilatõnõn iznini gerektirmez; İnternet adresi belirtilerek yayõn ve referans olarak kullanõlabilir. Bu e-kitap, http://ekutup.dpt.gov.tr/ adresindedir. Bu yayõn 325 adet basõlmõştõr. Elektronik olarak, 1 adet pdf dosyasõ üretilmiştir. http://ekutup.dpt.gov.tr/kit/cakalr/tekel.pdf Çakal Doğal Tekellerde Özelleştirme ve Regülasyon İÇİNDEKİLER Sayfa 1. GİRİŞ ........................................................................................................................................................................... 1 BİRİNCİ BÖLÜM EKONOMİK ETKİNLİK KAVRAMI VE EKONOMİDE DEVLET MÜDAHALESİNİN RASYONELİ 1. SERBEST REKABET PİYASALARI VE EKONOMİK ETKİNLİK............................................................. 5 1.1 EKONOMİK ETKİNLİK ......................................................................................................................................... 5 1.1.1 Tahsiste Etkinlik (Allocative Efficiency) ........................................................................................................... 6 1.1.2 Üretimde -

Action Needed: E-Cigarettes
ACTION NEEDED: E-CIGARETTES Since their introduction in 2008, e-cigarettes have become both ubiquitous and an increasing source of public policy concern and debate. This concern stems primarily from drastic increases in youth e-cigarette use. The most recent data show that 27.5% of high schoolers are using these products — a rate of youth tobacco product use not seen in nearly two decades. The public health community, parents and educators are shocked and worried to find their children and students using these products at home and even in class. Young people are reporting severe signs of dependence, E-cigarettes are now threatening including using e-cigarettes when they first wake up, inability to concentrate in the classroom without to undo all the progress that the using an e-cigarette, and even waking in the night public health community and to get a nicotine fix. government have made over decades We have known for decades that youth in particular to reduce cigarette smoking should not be exposed to nicotine because it changes brain chemistry to create a stronger addiction, can lead to memory and concentration Current generation of e-cigarettes problems, and can make youth who use it more susceptible to addiction to other substances. Research also suggests that young people who use e-cigarettes are four times more likely to go on to smoke combustible cigarettes. E-cigarettes are now threatening to undo all the progress that the public health community and JUUL government have made over decades to reduce cigarette smoking. Indeed, the surgeon general raised the alarm by issuing an advisory declaring a youth e-cigarette epidemic in December 2018. -

E-Cigarette Marketing Regulations and Youth Vaping: Cross-Sectional Surveys, 2017–2019 David Hammond, Phd, Jessica L
E-cigarette Marketing Regulations and Youth Vaping: Cross-Sectional Surveys, 2017–2019 David Hammond, PhD, Jessica L. Reid, MSc, Robin Burkhalter, MMath, Vicki L. Rynard, MSc BACKGROUND: Increased electronic cigarette (e-cigarette) use among young people is often abstract attributed to industry marketing practices; however, the effectiveness of regulations that limit e-cigarette advertising and promotions has yet to be examined. New federal legislation that liberalized the Canadian e-cigarette market in May 2018, along with differences in provincial regulations, provides an opportunity to examine the impact of regulatory restrictions on e-cigarette marketing. METHODS: Repeat cross-sectional surveys of 12 004 16- to 19-year-olds in Canada, completed online in 2017, 2018, and 2019. Logistic regression models were used to examine differences over time in exposure to e-cigarette marketing and e-cigarette use, including between provinces with differing strengths of marketing restrictions. RESULTS: The percentage of youth surveyed who reported noticing e-cigarette promotions often or very often approximately doubled between 2017 and 2019 (13.6% vs 26.0%; adjusted odds ratio [AOR] = 2.24, 95% confidence interval [CI] = 1.97–2.56). Overall exposure to marketing was generally more prevalent in provinces with fewer regulatory restrictions. Respondents who reported noticing marketing often or very often were more likely to report vaping in the past 30 days (AOR = 1.41, 95% CI = 1.23–1.62), past week (AOR = 1.44, 95% CI = 1.22–1.70), and $20 days in the past month (AOR = 1.42, 95% CI = 1.11–1.81, P = .005). Provinces with low restrictions on marketing had higher prevalence of vaping in the past 30 days (AOR = 1.50, 95% CI = 1.25–1.80, P , .001), and in the past week (AOR = 1.65, 95% CI = 1.33–2.05, P , .001). -

Engineering and Delivery of Synthetic Chromatin Effectors
Engineering and Delivery of Synthetic Chromatin Effectors by Stefan Josef Tekel A Dissertation Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy Approved March 2019 by the Graduate Supervisory Committee Karmella Haynes, Chair Michael Caplan Jeremy Mills David Brafman ARIZONA STATE UNIVERSITY May 2019 ABSTRACT Synthetic manipulation of chromatin dynamics has applications for medicine, agriculture, and biotechnology. However, progress in this area requires the identification of design rules for engineering chromatin systems. In this thesis, I discuss research that has elucidated the intrinsic properties of histone binding proteins (HBP), and apply this knowledge to engineer novel chromatin binding effectors. Results from the experiments described herein demonstrate that the histone binding domain from chromobox protein homolog 8 (CBX8) is portable and can be customized to alter its endogenous function. First, I developed an assay to identify engineered fusion proteins that bind histone post translational modifications (PTMs) in vitro and regulate genes near the same histone PTMs in living cells. This assay will be useful for assaying the function of synthetic histone PTM-binding actuators and probes. Next, I investigated the activity of a novel, dual histone PTM binding domain regulator called Pc2TF. I characterized Pc2TF in vitro and in cells and show it has enhanced binding and transcriptional activation compared to a single binding domain fusion called Polycomb Transcription Factor (PcTF). These results indicate that valency can be used to tune the activity of synthetic histone-binding transcriptional regulators. Then, I report the delivery of PcTF fused to a cell penetrating peptide (CPP) TAT, called CP-PcTF.