Comparative Phylogeography of Pine -Oak Birds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Groundwater Budgets for Detrital, Hualapai, and Sacramento Valleys, Mohave County, Arizona, 2007–08
Prepared in cooperation with the Arizona Department of Water Resources Groundwater Budgets for Detrital, Hualapai, and Sacramento Valleys, Mohave County, Arizona, 2007–08 Scientific Investigations Report 2011–5159 U.S. Department of the Interior U.S. Geological Survey COVER: View of August 2010, monsoon thunderstorm over Hualapai Mountains, looking east from Sacramento Valley. Photo by Bradley D. Garner. Prepared in cooperation with the Arizona Department of Water Resources Groundwater Budgets for Detrital, Hualapai, and Sacramento Valleys, Mohave County, Arizona, 2007–08 By Bradley D. Garner and Margot Truini Scientific Investigations Report 2011–5159 U.S. Department of the Interior U.S. Geological Survey U.S. Department of the Interior KEN SALAZAR, Secretary U.S. Geological Survey Marcia K. McNutt, Director U.S. Geological Survey, Reston, Virginia: 2011 For product and ordering information: World Wide Web: http://www.usgs.gov/pubprod Telephone: 1-888-ASK-USGS For more information on the USGS—the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment: World Wide Web: http://www.usgs.gov Telephone: 1-888-ASK-USGS Any use of trade, product, or firm names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Although this report is in the public domain, permission must be secured from the individual copyright owners to reproduce any copyrighted material contained within this report. Suggested citation: Garner, B.D., and Truini, Margot, 2011, Groundwater budgets for Detrital, Hualapai, and Sacramento Valleys, Mohave County, Arizona, 2007–08: U.S. Geological Survey Scientific Investigations Report 2011-5159, 34 p. -

Summits on the Air – ARM for the USA (W7A
Summits on the Air – ARM for the U.S.A (W7A - Arizona) Summits on the Air U.S.A. (W7A - Arizona) Association Reference Manual Document Reference S53.1 Issue number 5.0 Date of issue 31-October 2020 Participation start date 01-Aug 2010 Authorized Date: 31-October 2020 Association Manager Pete Scola, WA7JTM Summits-on-the-Air an original concept by G3WGV and developed with G3CWI Notice “Summits on the Air” SOTA and the SOTA logo are trademarks of the Programme. This document is copyright of the Programme. All other trademarks and copyrights referenced herein are acknowledged. Document S53.1 Page 1 of 15 Summits on the Air – ARM for the U.S.A (W7A - Arizona) TABLE OF CONTENTS CHANGE CONTROL....................................................................................................................................... 3 DISCLAIMER................................................................................................................................................. 4 1 ASSOCIATION REFERENCE DATA ........................................................................................................... 5 1.1 Program Derivation ...................................................................................................................................................................................... 6 1.2 General Information ..................................................................................................................................................................................... 6 1.3 Final Ascent -
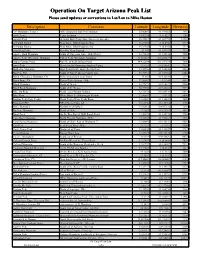
Peak List Please Send Updates Or Corrections to Lat/Lon to Mike Heaton
Operation On Target Arizona Peak List Please send updates or corrections to Lat/Lon to Mike Heaton Description Comment Latitude Longitude Elevation "A" Mountain (Tempe) ASU campus by Sun Devil Stadium 33.42801 -111.93565 1495 AAA Temp Temp Location 33.42234 -111.8227 1244 Agassiz Peak @ Snow Bowl Tram Stop (No access to peak) 35.32587 -111.67795 12353 Al Fulton Point 1 Near where SR260 tops the Rim 34.29558 -110.8956 7513 Al Fulton Point 2 Near where SR260 tops the rim 34.29558 -110.8956 7513 Alta Mesa Peak For Alta Mesa Sign-up 33.905 -111.40933 7128 Apache Maid Mountain South of Stoneman Lake - Hike/Drive? 34.72588 -111.55128 7305 Apache Peak, Whetstone Mountain Tallest Peak, Whetstone Mountain 31.824583 -110.429517 7711 Aspen Canyon Point Rim W. of Kehl Springs Point 34.422204 -111.337874 7600 Aztec Peak Sierra Ancha Mountains South of Young 33.8123 -110.90541 7692 Battleship Mountain High Point visible above the Flat Iron 33.43936 -111.44836 5024 Big Pine Flat South of Four Peaks on County Line 33.74931 -111.37304 6040 Black (Chocolate) Mountain, CA Drive up and park, near Yuma 33.055 -114.82833 2119 Black Butte, CA East of Palm Springs - Hike 33.56167 -115.345 4458 Black Mountain North of Oracle 32.77899 -110.96319 5586 Black Rock Mountain South of St. George 36.77305 -113.80802 7373 Blue Jay Ridge North end of Mount Graham 32.75872 -110.03344 8033 Blue Vista White Mtns. S. of Hannagan Medow 33.56667 -109.35 8000 Browns Peak (Four Peaks) North Peak of Four Peaks Range 33.68567 -111.32633 7650 Brunckow Hill NE of Sierra Vista, AZ 31.61736 -110.15788 4470 Bryce Mountain Northwest of Safford 33.02012 -109.67232 7298 Buckeye Mountain North of Globe 33.4262 -110.75763 4693 Burnt Point On the Rim East of Milk Ranch Point 34.40895 -111.20478 7758 Camelback Mountain North Phoenix Mountain - Hike 33.51463 -111.96164 2703 Carol Spring Mountain North of Globe East of Highway 77 33.66064 -110.56151 6629 Carr Peak S. -

Downloaded and Reviewed on the State Parks’ Webpage Or Those Interested Could Request a Hard Copy
Governor of Arizona Janet Napolitano Arizona State Parks Board William Cordasco, Chair ting 50 ting 50 ra Y Arlan Colton ra Y b e b e a William C. Porter a le le r r e e s s William C. Scalzo C C Tracey Westerhausen Mark Winkleman 1957 - 2007 Reese Woodling 1957 - 2007 Elizabeth Stewart (2006) Arizona Outdoor Recreation Coordinating Commission Jeffrey Bell, Chair Mary Ellen Bittorf Garry Hays Rafael Payan William Schwind Duane Shroufe Kenneth E. Travous This publication was prepared under the authority of the Arizona State Parks Board. Prepared by the Statewide Planning Unit Resources Management Section Arizona State Parks 1300 West Washington Street Phoenix, Arizona 85007 (602) 542-4174 Fax: (602) 542-4180 www.azstateparks.com The preparation of this report was under the guidance from the National Park Service, U.S. Department of the Interior, under the provisions of the Land and Water Conservation Fund Act of 1965 (Public Law 88-578, as amended). The Department of the Interior prohibits discrimination on the basis of race, religion, national origin, age or disability. For additional information or to file a discrimination complaint, contact Director, Office of Equal Opportunity, Department of the Interior, Washington D.C. 20240. September 2007 ARIZONA 2008 SCORP ARIZONA 2008 Statewide Comprehensive Outdoor Recreation Plan (SCORP) Arizona State Parks September 2007 iii ARIZONA 2008 SCORP ACKNOWLEDGEMENTS The 2008 Statewide Comprehensive Outdoor Recreation Plan (SCORP) for Arizona was prepared by the Planning Unit, Resources Management -

Grand Canyon Council Oa Where to Go Camping Guide
GRAND CANYON COUNCIL OA WHERE TO GO CAMPING GUIDE GRAND CANYON COUNCIL, BSA OA WHERE TO GO CAMPING GUIDE Table of Contents Introduction to The Order of the Arrow ....................................................................... 1 Wipala Wiki, The Man .................................................................................................. 1 General Information ...................................................................................................... 3 Desert Survival Safety Tips ........................................................................................... 4 Further Information ....................................................................................................... 4 Contact Agencies and Organizations ............................................................................. 5 National Forests ............................................................................................................. 5 U. S. Department Of The Interior - Bureau Of Land Management ................................ 7 Maricopa County Parks And Recreation System: .......................................................... 8 Arizona State Parks: .................................................................................................... 10 National Parks & National Monuments: ...................................................................... 11 Tribal Jurisdictions: ..................................................................................................... 13 On the Road: National -

FINAL Feasibility Report / Prepared for ADOT
FINAL FEASIBILITY REPORT / PREPARED FOR ADOT I-40/US 93 WEST KINGMAN TI Final Feasibility Report ADOT Project No. 040 M0 48 H732301L FEDERAL PROJECT NO. NH-040-A(AVJ) Topock-Kingman Highway I-40 October 2009 Prepared By Final Feasibility Report l-AÛlUS 93 West K¡ngman Tl ADOT PROJECT NO. O4O MO 48 H732301L FEDERAL PROJECT NO. NH-040-A (AVJ) TOPOCK - KINGMAN HIGHWAY t-40 Prepared for Arizona Department of Transportation October 2009 I-40/US 93 West Kingman TI Feasibility Report Executive Summary Background This study for the I-40/US 93 West Kingman TI, ADOT Project Number 040 MO 48 H732301L, identifies possible corridor alternatives for a new system-to-system directional interchange connecting Interstate 40 (I-40) to United States Route 93 (US 93) near Kingman, AZ. The general location for the various alternative corridors is along I-40 between the Shinarump Drive traffic interchange (TI) and Stockton Hill Road TI (approximate MP 44 to MP 52) and along US 93 between the State Route (SR) 68 TI, near the Kingman Port of Entry, and the existing Beale Street interchange, where US 93 connects with I-40 (approximate MP 67 to MP 71). US 93 is part of the CANAMEX Corridor. The Beale Street interchange between I-40 and US 93 is the third of three “bottleneck” locations along US 93 identified from the previous CANAMEX Corridor study. The first location is Hoover Dam and that area is being addressed with the Hoover Dam Bypass construction. The second location is Wickenburg and that area is being addressed with an interim bypass that began construction in February 2008. -

ARIZONA - BLM District and Field Office Boundaries
ARIZONA - BLM District and Field Office Boundaries Bea ve r Beaver Dam D r S Mountains e COLORADO CITY a a i v D m R (! Cottonwood Point sh RAINBOW LODGE u n a Wilderness C d (! I y W Paria Canyon - A W t ge S Sa GLEN CANYON z Y Cow Butte c A l A RED MESA h a a S Lake Powell t e k h n c h h te K Nokaito Bench ! El 5670 l ( s Vermilion Cliffs Mitchell Mesa a o C hi c S E d h S y a e u rt n W i n m Lost Spring Mountain Wilderness KAIByAo B- e s g u Coyote Butte RECREATION AREA O E h S C L r G H C n Wilderness a i l h FREDONIA r l a h ! r s V i ( N o re M C W v e (! s e m L (! n N l a o CANE BEDS a u l e a TES NEZ IAH W n MEXICAN WATER o k I s n k l A w W y a o M O N U M E N T (! W e GLEN CANYON DAM PAGE S C s A W T W G O c y V MOCCASIN h o k (! k W H a n R T Tse Tonte A o a El 5984 T n PAIUTE e n (! I N o E a N s t M y ES k h n s N e a T Meridian Butte l A o LITTLEFIELD c h I Mokaac Mountain PIPE SPRING e k M e o P A r d g R j o E n i (! J I A H e (! r A C r n d W l H a NATIONAL KAIBAB W U C E N k R a s E A h e i S S u S l d O R A c e e O A C a I C r l T r E MONIMENT A L Black Rock Point r t L n n i M M SWEETWATER r V A L L E Y i N c t N e (! a a h S Paiute U Vermilion Cliffs N.M. -

The Colorado's
HIKING WITH LLAMAS RELAXING IN A NORTH RIM CABIN arizonahighways.com JULY 2004 taste the colorado’s many moods Explore the Erratic Turns of the verde river Visit Our Western Waterworld parker strip See What Happened When They Tried to herd deer like cattle {also inside} 46 DESTINATION JULY 2004 Grand Canyon Lodge There are many exceptional places to visit and enjoy Arizona’s great Canyon, but the lodge on the North Rim has unequalled remoteness and coziness. 42 BACK ROAD ADVENTURE A drive from Munds Park to Mormon Lake recaptures the Old West spirit. 48 HIKE OF THE MONTH Five trails link up in Hualapai Mountain Park. Kaibab 2 LETTERS & E-MAIL Grand Canyon Plateau National Park Grand Canyon 3 TAKING THE Lodge OFF-RAMP Hualapai Mountain Park MUNDS Mormon Lake PARK Parker Strip STRAWBERRY 40 HUMOR Verde PARKER River Knoll Lake 41 ALONG THE WAY PHOENIX COVER/ADVENTURE RECREATION PORTFOLIO A family’s old green tent 6 32 18 was hard to leave behind Classic Wooden Boats Hiking With Llamas The Verde River — when the time came. TUCSON Near Strawberry, an overland trekking expedition with Ride the Big River Abundant and Glorious POINTS OF INTEREST the odd-but-lovable spitting critters proves humorous FEATURED IN THIS ISSUE A group of eccentric and fanatic admirers of old-style Central Arizona’s perennial waterway possesses and relaxing. wooden boat designs prove again that the vessels can multiple personalities — from calm to sometimes handle the Colorado River’s thundering white water. raging — and traverses historic lands where the state {more stories online} at arizonahighways.com TRAVEL was born. -

United States Department of the Interior National Park Service
NPS Form 10-900-b OMB No. 1024-0018 (March 1992) United States Department of the Interior National Park Service NATIONAL REGISTER OF HISTORIC PLACES MULTIPLE PROPERTY DOCUMENTATION FORM This form is used for documenting multiple property groups relating to one or several historic contexts. See instructions in How to Complete the Multiple Property Documentation Form (National Register Bulletin 16B). Complete each item by entering the requested information. For additional space, use continuation sheets (Form 10-900-a). Use a typewriter, word processor, or computer to complete all items. X New Submission Amended Submission ======================================================================== A. Name of Multiple Property Listing ================================================================================= Cattle Ranching in Arizona, 1540-1950 ======================================================================== B. Associated Historic Contexts ================================================================================= (Name each associated historic context, identifying theme, geographical area, and chronological period for each.) 1. Cattle in the Arizona Economy: An Overview. 2. The Spanish Colonial and Mexican Periods, 1521-1848. 3. The Early American Period, 1848-1880. 4. The Boom Period, 1880-1893. 5. The Foundation of the Modern Cattle Industry, 1893-1950. 6. Indian Cattle Raising: Case Studies of the San Carlos and Tohono O’odham Reservations, 1920-1950. 7. Southern Arizona Ranch House Architectural Context 8. Northern -

Mineral Resources of the Wabayuma Peak Wilderness Study Area, Mohave County, Arizona
Mineral Resources of the Wabayuma Peak Wilderness Study Area, Mohave County, Arizona U. S. GEOLOGICAL SURVEY BULLETIN 1 737- E CORRECTION FOR GPO JACKET (FY 91) 585048 (PRINT ORDER 30039) U.S. GEOLOGICAL SURVEY BULLETIN 1737-E SUP. DOCS.: This page is to be distributed according to the standard IGS mailing list for USGS bulletins. USER: On both plate 1 and figure 2: The dark-pink area in the northwestern part of the map is colored correctly in having high potential for deposit type [2]; however, it should not contain the horizontal-line pattern denoting moderate potential for deposit type [2], This horizontal-line pattern should also not appear in the narrow strip in the northwesternmost corner of the map area; this area has moderate potential for deposit type [3], as colored, but has no potential for deposit type [2]. On both plate 1 and figure 2, the pale-pink area in the southern and central part of the map is not correct; this area should contain only the horizontal-line pattern shown, denoting moderate potential for deposit type [2]. Chapter E Mineral Resources of the Wabayuma Peak Wilderness Study Area, Mohave County, Arizona By CLAY M. CONWAY, JERRY R. HASSEMER, DANIEL H. KNEPPER, JR., JAMES A. PITKIN, and ROBERT C. JACHENS U.S. Geological Survey MARK L. CHATMAN U.S. Bureau of Mines U.S. GEOLOGICAL SURVEY BULLETIN 1737 MINERAL RESOURCES OF WILDERNESS STUDY AREAS: BLACK MOUNTAINS REGION, ARIZONA U.S. DEPARTMENT OF THE INTERIOR MANUEL LUJAN, JR., Secretary U.S. GEOLOGICAL SURVEY Dallas L. Peck, Director Any use of trade, product, or firm names in this publication is for descriptive purposes only and does not imply endorsement by the U.S. -

United States Department of the Interior Bureau of Land Management
United States Department of the Interior Bureau of Land Management Environmental Assessment DOI-BLM-AZ-C010-2013-0018-EA Date March 05, 2013 Whiskey Basin Boundary Trail Maintenance U.S. Department of the Interior Bureau of Land Management Kingman Field Office 2755 Mission Blvd Kingman, AZ 86401 Phone: 928-718-3700 FAX: 928-718-3761 Title DOI-BLM-AZ-CO10-2013-0018-EA Whiskey Basin Boundary Trail Maintenance I. Introduction: The Whiskey Basin Boundary Trail Maintenance EA # DOI-BLM-AZ-C010-2013-0018- EA will be for the maintenance and rehabilitation of sections of off-highway vehicle (OHV) trails along the Wabayuma Peak Wilderness. Whiskey Basin Boundary Trail (Figure 1) is located in the Hualapai Mountains approximately 17 miles south of Kingman, east of Interstate Highway 40. The OHV trail is located in the Kingman Field Office (KFO) area of jurisdiction, located in T19N R16W sections 16, 21, and 28 of the Gila and Salt River Base Line and Meridian (USGS 7.5’ Quadrangle Hualapai Peak, Ariz.). A. Background: Wabayuma Peak area was designated as Wilderness with the passage of the Arizona Desert Wilderness Act of 1990. The Wabayuma Peak & Mount Tipton Wilderness Management Plan, Environmental Assessment, and Decision Record (WMP/EIS) was completed August 1995. Whiskey Basin is a natural bowl area providing outstanding views of the wilderness area and Wabayuma Peak and is a popular four-wheel drive destination. This OHV trail provides access to five hiking trails in the Wabayuma Peak Wilderness. B. Purpose and Need: The purpose of the proposed action is to comply with the policy statement of the Federal Land Policy and Management Act of 1976 (Sec. -

Bill Williams River Watershed 5,393 52% 48%
NEMO Watershed Based Plan Bill Williams Watershed Acknowledgements Arizona NEMO acknowledges the University of Arizona Cooperative Extension Service, Arizona Department of Environmental Quality (ADEQ) Water Quality Division, the Water Resources Research Center, and the University of Arizona Advanced Resource Technology Lab (ART) for their technical support in producing the Watershed Based Plans. Funding provided by the U.S. Environmental Protection Agency under the Clean Water Act and the Arizona Department of Environmental Quality’s Water Quality Protection Division. Additional financial support is provided by the University of Arizona, Technology and Research Initiative Fund (TRIF), Water Sustainability Program through the Water Resources Research Center. The NEMO website is www.ArizonaNEMO.org. Written and prepared by: Hoori Ajami, D. Phillip Guertin, Lainie R. Levick and Kristine Uhlman University of Arizona Tucson, Arizona December 2005 Table of Contents Section 1: Introduction Background Purpose and Scope Methods Structure of this Watershed Plan References Section 2: Physical Features Watershed Size Topography Water Resources Lakes and Reservoirs Stream Type Stream Density Annual Stream Flow Water Quality Geology Soils Climate Precipitation Temperature References Data Sources Section 3: Biological Resources Ecoregions Vegetation Habitats (Riparian and Wetland Areas) Major Land Resource Areas (MLRAs) References Data Sources Section 4: Social / Economic Characteristics County Governments Council of Governments (COGs) Urban Areas Roads