ASH2L Drives Proliferation and Sensitivity to Bleomycin and Other
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin
cells Review Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin Agnieszka Bochy ´nska,Juliane Lüscher-Firzlaff and Bernhard Lüscher * ID Institute of Biochemistry and Molecular Biology, Medical School, RWTH Aachen University, Pauwelsstrasse 30, 52057 Aachen, Germany; [email protected] (A.B.); jluescher-fi[email protected] (J.L.-F.) * Correspondence: [email protected]; Tel.: +49-241-8088850; Fax: +49-241-8082427 Received: 18 January 2018; Accepted: 27 February 2018; Published: 2 March 2018 Abstract: Regulation of gene expression is achieved by sequence-specific transcriptional regulators, which convey the information that is contained in the sequence of DNA into RNA polymerase activity. This is achieved by the recruitment of transcriptional co-factors. One of the consequences of co-factor recruitment is the control of specific properties of nucleosomes, the basic units of chromatin, and their protein components, the core histones. The main principles are to regulate the position and the characteristics of nucleosomes. The latter includes modulating the composition of core histones and their variants that are integrated into nucleosomes, and the post-translational modification of these histones referred to as histone marks. One of these marks is the methylation of lysine 4 of the core histone H3 (H3K4). While mono-methylation of H3K4 (H3K4me1) is located preferentially at active enhancers, tri-methylation (H3K4me3) is a mark found at open and potentially active promoters. Thus, H3K4 methylation is typically associated with gene transcription. The class 2 lysine methyltransferases (KMTs) are the main enzymes that methylate H3K4. KMT2 enzymes function in complexes that contain a necessary core complex composed of WDR5, RBBP5, ASH2L, and DPY30, the so-called WRAD complex. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis
International Journal of Molecular Sciences Review Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis Elzbieta Poreba 1,* , Krzysztof Lesniewicz 2 and Julia Durzynska 1,* 1 Institute of Experimental Biology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland 2 Department of Molecular and Cellular Biology, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland; [email protected] * Correspondence: [email protected] (E.P.); [email protected] (J.D.); Tel.: +48-61-829-5857 (E.P.) Received: 19 November 2020; Accepted: 6 December 2020; Published: 8 December 2020 Abstract: KMT2 (histone-lysine N-methyltransferase subclass 2) complexes methylate lysine 4 on the histone H3 tail at gene promoters and gene enhancers and, thus, control the process of gene transcription. These complexes not only play an essential role in normal development but have also been described as involved in the aberrant growth of tissues. KMT2 mutations resulting from the rearrangements of the KMT2A (MLL1) gene at 11q23 are associated with pediatric mixed-lineage leukemias, and recent studies demonstrate that KMT2 genes are frequently mutated in many types of human cancers. Moreover, other components of the KMT2 complexes have been reported to contribute to oncogenesis. This review summarizes the recent advances in our knowledge of the role of KMT2 complexes in cell transformation. In addition, it discusses the therapeutic targeting of different components of the KMT2 complexes. Keywords: histone–lysine N-methyltransferase 2; COMPASS; COMPASS-like; H3K4 methylation; oncogenesis; cancer; epigenetics; chromatin 1. -

EBF1 Drives Hallmark B Cell Gene Expression by Enabling the Interaction of PAX5 with the MLL H3K4 Methyltransferase Complex Charles E
www.nature.com/scientificreports OPEN EBF1 drives hallmark B cell gene expression by enabling the interaction of PAX5 with the MLL H3K4 methyltransferase complex Charles E. Bullerwell1, Philippe Pierre Robichaud1,2, Pierre M. L. Deprez1, Andrew P. Joy1, Gabriel Wajnberg1, Darwin D’Souza1,3, Simi Chacko1, Sébastien Fournier1, Nicolas Crapoulet1, David A. Barnett1,2, Stephen M. Lewis1,2 & Rodney J. Ouellette1,2* PAX5 and EBF1 work synergistically to regulate genes that are involved in B lymphocyte diferentiation. We used the KIS-1 difuse large B cell lymphoma cell line, which is reported to have elevated levels of PAX5 expression, to investigate the mechanism of EBF1- and PAX5-regulated gene expression. We demonstrate the lack of expression of hallmark B cell genes, including CD19, CD79b, and EBF1, in the KIS-1 cell line. Upon restoration of EBF1 expression we observed activation of CD19, CD79b and other genes with critical roles in B cell diferentiation. Mass spectrometry analyses of proteins co-immunoprecipitated with PAX5 in KIS-1 identifed components of the MLL H3K4 methylation complex, which drives histone modifcations associated with transcription activation. Immunoblotting showed a stronger association of this complex with PAX5 in the presence of EBF1. Silencing of KMT2A, the catalytic component of MLL, repressed the ability of exogenous EBF1 to activate transcription of both CD19 and CD79b in KIS-1 cells. We also fnd association of PAX5 with the MLL complex and decreased CD19 expression following silencing of KMT2A in other human B cell lines. These data support an important role for the MLL complex in PAX5-mediated transcription regulation. -

Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal Through Mirna-1247-3P in Human Cervical Cancer Cells
Article Volume 11, Issue 1, 2021, 7943 - 7972 https://doi.org/10.33263/BRIAC111.79437972 Anticancer Effect of Natural Product Sulforaphane by Targeting MAPK Signal through miRNA-1247-3p in Human Cervical Cancer Cells Meng Luo 1 , Dian Fan 2 , Yinan Xiao 2 , Hao Zeng 2 , Dingyue Zhang 2 , Yunuo Zhao 2 , Xuelei Ma 1,* 1 Department of Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (L.M.); 2 West China Medical School, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China; [email protected] (F.D.); [email protected] (X.Y.N.); [email protected] (Z.H.); [email protected] (Z.D.Y.); [email protected] (Z.Y.N.); * Correspondence: [email protected]; Scopus Author ID 55523405000 Received: 11.06.2020; Revised: 3.07.2020; Accepted: 5.07.2020; Published: 9.07.2020 Abstract: The prognosis of cervical cancer remains poor. Sulforaphane, an active ingredient from cruciferous plants, has been identified as a potential anticancer agent in various cancers. However, there is little information about its effect on cervical cancer. Here, we conducted a present study to uncover the effect and the potential mechanisms of sulforaphane on cervical cancer. HeLa cells were treated with sulforaphane, and cell proliferation and apoptosis were assessed by Cell Counting Kit-8, Western blot, flow cytometry, and immunofluorescence. Then, next-generation sequencing (NGS) and bioinformatics tools were used to analyze mRNA-seq, miRNA-seq, and potential pathways. Finally, qRT-PCR, Cell Counting Kit-8, flow cytometry, small RNAs analysis, and Western blot were performed to evaluate the biological function of miR1247-3p and MAPK pathway in HeLa cell lines. -

The H3K4 Methyltransferase Setd1b Is Essential for Hematopoietic Stem and Progenitor Cell Homeostasis in Mice
RESEARCH ARTICLE The H3K4 methyltransferase Setd1b is essential for hematopoietic stem and progenitor cell homeostasis in mice Kerstin Schmidt1, Qinyu Zhang2, Alpaslan Tasdogan3,4, Andreas Petzold5, Andreas Dahl5, Borros M Arneth6, Robert Slany7, Hans Jo¨ rg Fehling3, Andrea Kranz2*, Adrian Francis Stewart2*, Konstantinos Anastassiadis1* 1Stem Cell Engineering, Biotechnology Center, Technische Universita¨ t Dresden, Dresden, Germany; 2Genomics, Biotechnology Center, Technische Universita¨ t Dresden, Dresden, Germany; 3Institute of Immunology, University Hospital Ulm, Ulm, Germany; 4Department of Dermatology, University Hospital Ulm, Ulm, Germany; 5Deep Sequencing Group, DFG - Center for Regenerative Therapies Dresden, Dresden, Germany; 6Institute of Laboratory Medicine and Pathobiochemistry, Molecular Diagnostics, Hospital of the Universities Giessen and Marburg, Giessen, Germany; 7Department of Genetics, Friedrich Alexander Universita¨ t Erlangen, Erlangen, Germany Abstract Hematopoietic stem cells require MLL1, which is one of six Set1/Trithorax-type histone 3 lysine 4 (H3K4) methyltransferases in mammals and clinically the most important leukemia gene. Here, we add to emerging evidence that all six H3K4 methyltransferases play essential roles in the hematopoietic system by showing that conditional mutagenesis of Setd1b in adult mice provoked *For correspondence: aberrant homeostasis of hematopoietic stem and progenitor cells (HSPCs). Using both ubiquitous [email protected] (AK); and hematopoietic-specific deletion strategies, the loss of Setd1b resulted in peripheral thrombo- [email protected] and lymphocytopenia, multilineage dysplasia, myeloid-biased extramedullary hematopoiesis in the (AFS); spleen, and lethality. By transplantation experiments and expression profiling, we determined that konstantinos.anastassiadis@tu- Setd1b is autonomously required in the hematopoietic lineages where it regulates key lineage dresden.de (KA) specification components, including Cebpa, Gata1, and Klf1. -
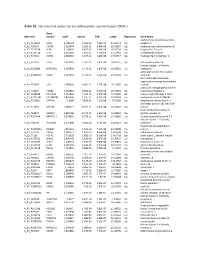
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454 -

Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase
life Review Structure, Activity and Function of the MLL2 (KMT2B) Protein Lysine Methyltransferase Alexia Klonou 1, Sarantis Chlamydas 1,2 and Christina Piperi 1,* 1 Department of Biological Chemistry, Medical School, National and Kapodistrian University of Athens, 11527 Athens, Greece; [email protected] (A.K.); [email protected] (S.C.) 2 Research and Development Department, Active Motif, Inc., Carlsbad, CA 92008, USA * Correspondence: [email protected]; Tel.: +30-210-7462610 Abstract: The Mixed Lineage Leukemia 2 (MLL2) protein, also known as KMT2B, belongs to the fam- ily of mammalian histone H3 lysine 4 (H3K4) methyltransferases. It is a large protein of 2715 amino acids, widely expressed in adult human tissues and a paralog of the MLL1 protein. MLL2 contains a characteristic C-terminal SET domain responsible for methyltransferase activity and forms a protein complex with WRAD (WDR5, RbBP5, ASH2L and DPY30), host cell factors 1/2 (HCF 1/2) and Menin. The MLL2 complex is responsible for H3K4 trimethylation (H3K4me3) on specific gene promoters and nearby cis-regulatory sites, regulating bivalent developmental genes as well as stem cell and germinal cell differentiation gene sets. Moreover, MLL2 plays a critical role in development and germ line deletions of Mll2 have been associated with early growth retardation, neural tube defects and apoptosis that leads to embryonic death. It has also been involved in the control of voluntary movement and the pathogenesis of early stage childhood dystonia. Additionally, tumor-promoting functions of MLL2 have been detected in several cancer types, including colorectal, hepatocellu- lar, follicular cancer and gliomas. In this review, we discuss the main structural and functional Citation: Klonou, A.; Chlamydas, S.; aspects of the MLL2 methyltransferase with particular emphasis on transcriptional mechanisms, Piperi, C. -

Neuronal Kmt2a/Mll1 Histone Methyltransferase Is Essential for Prefrontal Synaptic Plasticity and Working Memory
The Journal of Neuroscience, April 1, 2015 • 35(13):5097–5108 • 5097 Neurobiology of Disease Neuronal Kmt2a/Mll1 Histone Methyltransferase Is Essential for Prefrontal Synaptic Plasticity and Working Memory Mira Jakovcevski,1,6* Hongyu Ruan,3* Erica Y. Shen,1* XAslihan Dincer,1,2* Behnam Javidfar,1 Qi Ma,2 XCyril J. Peter,1 Iris Cheung,4 Amanda C. Mitchell,3 Yan Jiang,1 Cong L. Lin,1 Venu Pothula,1 A. Francis Stewart,7 Patricia Ernst,5 Wei-Dong Yao,3 and Schahram Akbarian1,4 1Department of Psychiatry and Friedman Brain Institute and 2Department of Genetics and Genomics Science and Institute for Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, New York 10029, 3New England Primate Center, Harvard Medical School, Southborough, Massachusetts 01702, 4Brudnick Neuropsychiatric Research Institute, University of Massachusetts Medical School, Worcester, Massachusetts 01604, 5Department of Genetics and Department of Microbiology and Immunology, Norris Cotton Cancer Center, Geisel School of Medicine at Dartmouth, Hanover New Hampshire 03755, 6Max Planck Institute of Psychiatry, 80804 Munich, Germany, and 7Genomics BioinnovationsZentrum, Technische Universitaet, 01307 Dresden, Germany Neuronal histone H3-lysine 4 methylation landscapes are defined by sharp peaks at gene promoters and other cis-regulatory sequences, but molecular and cellular phenotypes after neuron-specific deletion of H3K4 methyl-regulators remain largely unexplored. We report thatneuronalablationoftheH3K4-specificmethyltransferase,Kmt2a/Mixed-lineageleukemia1(Mll1),inmousepostnatalforebrainand adult prefrontal cortex (PFC) is associated with increased anxiety and robust cognitive deficits without locomotor dysfunction. In contrast, only mild behavioral phenotypes were observed after ablation of the Mll1 ortholog Kmt2b/Mll2 in PFC. Impaired working memory after Kmt2a/Mll1 ablation in PFC neurons was associated with loss of training-induced transient waves of Arc immediate early gene expression critical for synaptic plasticity. -

Epigenome Alterations in Aortic Valve Stenosis and Its Related Left
Gošev et al. Clinical Epigenetics (2017) 9:106 DOI 10.1186/s13148-017-0406-7 REVIEW Open Access Epigenome alterations in aortic valve stenosis and its related left ventricular hypertrophy Igor Gošev1, Martina Zeljko2, Željko Đurić3, Ivana Nikolić4, Milorad Gošev5, Sanja Ivčević6, Dino Bešić7, Zoran Legčević7 and Frane Paić7* Abstract Aortic valve stenosis is the most common cardiac valve disease, and with current trends in the population demographics, its prevalence is likely to rise, thus posing a major health and economic burden facing the worldwide societies. Over the past decade, it has become more than clear that our traditional genetic views do not sufficiently explain the well-known link between AS, proatherogenic risk factors, flow-induced mechanical forces, and disease-prone environmental influences. Recent breakthroughs in the field of epigenetics offer us a new perspective on gene regulation, which has broadened our perspective on etiology of aortic stenosis and other aortic valve diseases. Since all known epigenetic marks are potentially reversible this perspective is especially exciting given the potential for development of successful and non-invasive therapeutic intervention and reprogramming of cells at the epigenetic level even in the early stages of disease progression. This review will examine the known relationships between four major epigenetic mechanisms: DNA methylation, posttranslational histone modification, ATP-dependent chromatin remodeling, and non-coding regulatory RNAs, and initiation and progression of AS. Numerous profiling and functional studies indicate that they could contribute to endothelial dysfunctions, disease-prone activation of monocyte-macrophage and circulatory osteoprogenitor cells and activation and osteogenic transdifferentiation of aortic valve interstitial cells, thus leading to valvular inflammation, fibrosis, and calcification, and to pressure overload-induced maladaptive myocardial remodeling and left ventricular hypertrophy. -

Long Non-Coding RNA FENDRR: Gene Structure, Expression, and Biological Relevance
G C A T T A C G G C A T genes Review Long Non-Coding RNA FENDRR: Gene Structure, Expression, and Biological Relevance Przemyslaw Szafranski * and Paweł Stankiewicz Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; [email protected] * Correspondence: [email protected] Abstract: The FOXF1 Adjacent Noncoding Developmental Regulatory RNA (Fendrr) plays an impor- tant role in the control of gene expression in mammals. It is transcribed in the opposite direction to the neighboring Foxf1 gene with which it shares a region containing promoters. In humans, FENDRR is located on chromosome 16q24.1, and is positively regulated both by the FOXF1 distant lung-specific cis-acting enhancer and by trans-acting FOXF1. Fendrr has been shown to function as a competing endogenous RNA, sponging microRNAs and protein factors that control stability of mRNAs, and as an epigenetic modifier of chromatin structure around gene promoters and other regulatory sites, targeting them with histone methyltrasferase complexes. In mice, Fendrr is essential for development of the heart, lungs, and gastrointestinal system; its homozygous loss causes embryonic or perina- tal lethality. Importantly, deregulation of FENDRR expression has been causatively linked also to tumorigenesis, resistance to chemotherapy, fibrosis, and inflammatory diseases. Here, we review the current knowledge on the FENDRR structure, expression, and involvement in development and tissue maintenance. Keywords: regulatory RNA; ceRNA; divergent genes; lncRNA enhancer; lung development; fibrosis; drug resistance Citation: Szafranski, P.; Stankiewicz, P. Long Non-Coding RNA FENDRR: Gene Structure, Expression, and Biological Relevance. Genes 2021, 12, 1. Introduction 177. https://doi.org/10.3390/genes 12020177 Nearly the entire human genome is transcribed; however, only approximately 2% of the transcriptome becomes translated into polypeptides longer than 100 amino acids [1,2]. -

You Can Check If Genes Are Captured by the Agilent Sureselect V5 Exome
IIHG Clinical Exome Sequencing Test Gene Coverage • Whole Exome Sequencing is a targeted capture platform which does not capture the entire exome. Regions not captured by the exome will not be analyzed. o Please note, it is important to understand the absence of a reportable variant in a given gene does not mean there are not pathogenic variants in that gene. • Data sensitivity and specificity for exome testing is variable as gene coverage is not uniform throughout the exome. • The Agilent SureSelect XT Human All Exon v5 kit captures ~98% of Refseq coding base pairs, and >94% of the captured coding bases in the exome are covered at our depth-of-coverage minimum threshold (30 reads). • All test reports include the following information: o Regions of the Symptom Candidate Gene List which were not captured by the current exome capture platform. o Regions of the Symptom Candidate Gene List which were not sequenced to a sufficient depth of coverage to make a clinical diagnosis. • You can check if genes are captured by the Agilent Agilent SureSelect v5 exome capture, and how well those genes are typically covered here. • Explanations for the headings; • Symbol: Gene symbol • Chr: Chromosome • % Captured: the minimum portion of the gene captured by the Agilent SureSelect XT Human All Exon v5 kit. This includes all transcripts for a given gene. o 100.00% = the entire gene is captured o 0.00% = none of the gene is captured • % Covered: the minimum portion of the gene that has at least 30x average coverage when sequenced on the Illumina HiSeq2000 with 100 base pair (bp) paired-end reads for eight CEPH samples.