The Conserved ASTN2/BRINP1 Locus at 9Q33.1–33.2 Is Associated With
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Genome-Wide Association Study Lystra P
Hayden et al. Respiratory Research (2018) 19:209 https://doi.org/10.1186/s12931-018-0890-0 RESEARCH Open Access Childhood asthma is associated with COPD and known asthma variants in COPDGene: a genome-wide association study Lystra P. Hayden1,2* , Michael H. Cho2,3, Benjamin A. Raby1,2,3, Terri H. Beaty4, Edwin K. Silverman2,3, Craig P. Hersh2,3 and on behalf of the COPDGene Investigators Abstract Background: Childhood asthma is strongly influenced by genetics and is a risk factor for reduced lung function and chronic obstructive pulmonary disease (COPD) in adults. This study investigates self-reported childhood asthma in adult smokers from the COPDGene Study. We hypothesize that childhood asthma is associated with decreased lung function, increased risk for COPD, and that a genome-wide association study (GWAS) will show association with established asthma variants. Methods: We evaluated current and former smokers ages 45–80 of non-Hispanic white (NHW) or African American (AA) race. Childhood asthma was defined by self-report of asthma, diagnosed by a medical professional, with onset at < 16 years or during childhood. Subjects with a history of childhood asthma were compared to those who never had asthma based on lung function, development of COPD, and genetic variation. GWAS was performed in NHW and AA populations, and combined in meta-analysis. Two sets of established asthma SNPs from published literature were examined for association with childhood asthma. Results: Among 10,199 adult smokers, 730 (7%) reported childhood asthma and 7493 (73%) reported no history of asthma. Childhood asthmatics had reduced lung function and increased risk for COPD (OR 3.42, 95% CI 2.81–4.18). -

Breakout Room 5 Poster Presentation Event
Akshara Chandrabalan 2020 NSERC USRA Bioresource Engineering, Supervisor Dr. Prasher 2020 Undergraduate Student Research Awards Predicting Biochar Sorption Capacities Using VIEW THE POSTERS Artificial Neural Networks Poster Presentation Event Breakout Room 5 Daniel Moses 2020 NSERC USRA (ZOOM) SEPTEMBER 17, 2020 @ 4pm Parasitology, Supervisor Dr. Salavati In search of the cryptic motif VI on trypanosomatid RNA editing ligases Richard Boivin 2020 NSERC USRA Antoine Gaudreau 2020 NSERC USRA 1 Natural Resource Sciences, Supervisor Dr. Humphries Bioresource Engineering, Supervisor Dr. Akbarzadeh Muriel Wong Min 2020 AES Brown Martlet USRA Evaluating regional patterns of traditional wildlife 3D Printed Agricultural Wastes for Advanced Food Science & Ag. Chemistry, Supervisor Dr. Karboune harvest in northern Quebec Biocomposites A Database Platform for The Selection of Appropriate Fat Substitutes as "Natural" Katerina Lazaris 2020 NSERC USRA Rowena Groeneveld 2020 NSERC USRA Food Ingredients Animal Science, Supervisor Dr. Bordignon Parasitology, Supervisor Dr. Beech Review: Methods to Improve Oocyte Competence and Modelling of a ligand gated ion channel from embryo Development in Prepubertal Animals to Caenorhabditis elegans experimentally shown to bind Liana Fortin-Hamel 2020 NSERC USRA Accerlerate genetic Gain 6 dopamine Parasitology, Supervisor Dr. Scott 2 Trisha Sackey 2020 NSERC USRA Effects of maternal nematode infection on spatial Food Science & Ag.Chemistry, Supervisor Dr. George Alex Zvezdin 2020 NSERC USRA learning and memory of young mouse pups Natural Resource Sciences, Supervisor Dr. Head Genotypic and Phenotypic Profiling of Staphylo- coccus aureus strains isolated from Canadian Impacts of Agricultural Practices on Yellow Perch Xavier Godin 2020 NSERC USRA Dairy Cattle for Antimicrobial Resistance Spawning Sites in Lac-Saint Pierre Québec Plant Science, Supervisor Dr. -

Investigating Developmental and Functional Deficits in Neurodegenerative
UNIVERSITY OF CALIFORNIA, IRVINE Investigating developmental and functional deficits in neurodegenerative disease using transcriptomic analyses DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences by Ryan Gar-Lok Lim Dissertation Committee: Professor Leslie M. Thompson, Chair Assistant Professor Dritan Agalliu Professor Peter Donovan Professor Suzanne Sandmeyer 2016 Introduction, Figure 1.1 © 2014 Macmillan Publishers Limited. Appendix 1 © 2016 Elsevier Ltd. All other materials © 2016 Ryan Gar-Lok Lim DEDICATION This dissertation is dedicated to my parents, sister, and my wife. I love you all very much and could not have accomplished any of this without your love and support. Please take the time to reflect back on all of the moments we’ve shared, and know, that it is because of those moments I have been able to succeed. This accomplishment is as much yours as it is mine. ii TABLE OF CONTENTS Page LIST OF FIGURES vi LIST OF TABLES ix ACKNOWLEDGMENTS x CURRICULUM VITAE xiii ABSTRACT OF THE DISSERTATION xv Introduction Huntington’s disease, the neurovascular unit and the blood-brain barrier 1 1.1 Huntington’s Disease 1.2 HTT structure and function 1.2.1 Normal HTT function and possible loss-of-function contributions to HD 1.3 mHTT pathogenesis 1.3.1 The dominant pathological features of mHTT - a gain-of- toxic function? 1.3.2 Cellular pathologies and non-neuronal contributions to HD 1.4 The neurovascular unit and the blood-brain barrier 1.4.1 Structure and function -

The Database of Chromosome Imbalance Regions and Genes
Lo et al. BMC Cancer 2012, 12:235 http://www.biomedcentral.com/1471-2407/12/235 RESEARCH ARTICLE Open Access The database of chromosome imbalance regions and genes resided in lung cancer from Asian and Caucasian identified by array-comparative genomic hybridization Fang-Yi Lo1, Jer-Wei Chang1, I-Shou Chang2, Yann-Jang Chen3, Han-Shui Hsu4, Shiu-Feng Kathy Huang5, Fang-Yu Tsai2, Shih Sheng Jiang2, Rajani Kanteti6, Suvobroto Nandi6, Ravi Salgia6 and Yi-Ching Wang1* Abstract Background: Cancer-related genes show racial differences. Therefore, identification and characterization of DNA copy number alteration regions in different racial groups helps to dissect the mechanism of tumorigenesis. Methods: Array-comparative genomic hybridization (array-CGH) was analyzed for DNA copy number profile in 40 Asian and 20 Caucasian lung cancer patients. Three methods including MetaCore analysis for disease and pathway correlations, concordance analysis between array-CGH database and the expression array database, and literature search for copy number variation genes were performed to select novel lung cancer candidate genes. Four candidate oncogenes were validated for DNA copy number and mRNA and protein expression by quantitative polymerase chain reaction (qPCR), chromogenic in situ hybridization (CISH), reverse transcriptase-qPCR (RT-qPCR), and immunohistochemistry (IHC) in more patients. Results: We identified 20 chromosomal imbalance regions harboring 459 genes for Caucasian and 17 regions containing 476 genes for Asian lung cancer patients. Seven common chromosomal imbalance regions harboring 117 genes, included gain on 3p13-14, 6p22.1, 9q21.13, 13q14.1, and 17p13.3; and loss on 3p22.2-22.3 and 13q13.3 were found both in Asian and Caucasian patients. -

Molecular Analyses of Malignant Pleural Mesothelioma
Molecular Analyses of Malignant Pleural Mesothelioma Shir Kiong Lo National Heart and Lung Institute Imperial College Dovehouse Street London SW3 6LY A thesis submitted for MD (Res) Faculty of Medicine, Imperial College London 2016 1 Abstract Malignant pleural mesothelioma (MPM) is an aggressive cancer that is strongly associated with asbestos exposure. Majority of patients with MPM present with advanced disease and the treatment paradigm mainly involves palliative chemotherapy and best supportive care. The current chemotherapy options are limited and ineffective hence there is an urgent need to improve patient outcomes. This requires better understanding of the genetic alterations driving MPM to improve diagnostic, prognostic and therapeutic strategies. This research aims to gain further insights in the pathogenesis of MPM by exploring the tumour transcriptional and mutational profiles. We compared gene expression profiles of 25 MPM tumours and 5 non-malignant pleura. This revealed differentially expressed genes involved in cell migration, invasion, cell cycle and the immune system that contribute to the malignant phenotype of MPM. We then constructed MPM-associated co-expression networks using weighted gene correlation network analysis to identify clusters of highly correlated genes. These identified three distinct molecular subtypes of MPM associated with genes involved in WNT and TGF-ß signalling pathways. Our results also revealed genes involved in cell cycle control especially the mitotic phase correlated significantly with poor prognosis. Through exome analysis of seven paired tumour/blood and 29 tumour samples, we identified frequent mutations in BAP1 and NF2. Additionally, the mutational profile of MPM is enriched with genes encoding FAK, MAPK and WNT signalling pathways. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Regulation of gene expression programs by serum response factor and megakaryoblastic leukemia 1/2 in macrophages Permalink https://escholarship.org/uc/item/8cc7d0t0 Author Sullivan, Amy Lynn Publication Date 2009 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Regulation of Gene Expression Programs by Serum Response Factor and Megakaryoblastic Leukemia 1/2 in Macrophages A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Amy Lynn Sullivan Committee in charge: Professor Christopher K. Glass, Chair Professor Stephen M. Hedrick Professor Marc R. Montminy Professor Nicholas J. Webster Professor Joseph L. Witztum 2009 Copyright Amy Lynn Sullivan, 2009 All rights reserved. The Dissertation of Amy Lynn Sullivan is approved, and it is acceptable in quality and form for publication on microfilm and electronically: ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ ______________________________________________________________ Chair University of California, San Diego 2009 iii DEDICATION To my husband, Shane, for putting up with me through all of the long hours, last minute late nights, and for not letting me quit no matter how many times my projects fell apart. To my son, Tyler, for always making me smile and for making every day an adventure. To my gifted colleagues, for all of the thought-provoking discussions, technical help and moral support through the roller- coaster ride that has been my graduate career. To my family and friends, for all of your love and support. I couldn’t have done it without you! iv EPIGRAPH If at first you don’t succeed, try, try, again. -

Peripherally Generated Foxp3+ Regulatory T Cells Mediate the Immunomodulatory Effects of Ivig in Allergic Airways Disease
Published February 20, 2017, doi:10.4049/jimmunol.1502361 The Journal of Immunology Peripherally Generated Foxp3+ Regulatory T Cells Mediate the Immunomodulatory Effects of IVIg in Allergic Airways Disease Amir H. Massoud,*,†,1 Gabriel N. Kaufman,* Di Xue,* Marianne Be´land,* Marieme Dembele,* Ciriaco A. Piccirillo,‡ Walid Mourad,† and Bruce D. Mazer* IVIg is widely used as an immunomodulatory therapy. We have recently demonstrated that IVIg protects against airway hyper- responsiveness (AHR) and inflammation in mouse models of allergic airways disease (AAD), associated with induction of Foxp3+ regulatory T cells (Treg). Using mice carrying a DTR/EGFP transgene under the control of the Foxp3 promoter (DEREG mice), we demonstrate in this study that IVIg generates a de novo population of peripheral Treg (pTreg) in the absence of endogenous Treg. IVIg-generated pTreg were sufficient for inhibition of OVA-induced AHR in an Ag-driven murine model of AAD. In the absence of endogenous Treg, IVIg failed to confer protection against AHR and airway inflammation. Adoptive transfer of purified IVIg-generated pTreg prior to Ag challenge effectively prevented airway inflammation and AHR in an Ag-specific manner. Microarray gene expression profiling of IVIg-generated pTreg revealed upregulation of genes associated with cell cycle, chroma- tin, cytoskeleton/motility, immunity, and apoptosis. These data demonstrate the importance of Treg in regulating AAD and show that IVIg-generated pTreg are necessary and sufficient for inhibition of allergen-induced AAD. The ability of IVIg to generate pure populations of highly Ag-specific pTreg represents a new avenue to study pTreg, the cross-talk between humoral and cellular immunity, and regulation of the inflammatory response to Ags. -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

The Role of ARHGAP9: Clinical Implication and Potential Function In
Han et al. J Transl Med (2021) 19:65 https://doi.org/10.1186/s12967-021-02733-5 Journal of Translational Medicine RESEARCH Open Access The role of ARHGAP9: clinical implication and potential function in acute myeloid leukemia Caixia Han1, Shujiao He1, Ruiqi Wang2, Xuefeng Gao1, Hong Wang2, Jingqiao Qiao1, Xiangyu Meng1, Yonghui Li1* and Li Yu1* Abstract Background: Rho GTPase activating protein 9 (ARHGAP9) is expressed in various types of cancers and can inactivate Rho GTPases that mainly regulate cytoskeletal dynamics. However, the exact role of ARHGAP9 in acute myeloid leuke- mia (AML) has yet to be clarifed. Methods: We compared the transcriptional expression, prognosis, diferentially expressed genes, functional enrich- ment, and hub genes in AML patients on the basis of the data published in the following databases: UALCAN, GEPIA, Gene Expression Omnibus, the Human Protein Atlas, Cancer Cell Line Encyclopedia, LinkedOmics, Metascape, and String. Data from the Cancer Genome Atlas database was used to evaluate the correlations between ARHGAP9 expression and various clinicopathological parameters, as well as the signifcantly diferent genes associated with ARHGAP9 expression. Results: We found that ARHGAP9 expression was higher in the tissues and cell lines extracted from patients with AML than corresponding control tissues and other cancer types. ARHGAP9 overexpression was associated with decreased overall survival (OS) in AML. Compared with the ARHGAP9low group, the ARHGAP9high group, which received only chemotherapy, showed signifcantly worse OS and event-free survival (EFS); however, no signifcant diference was observed after treatment with autologous or allogeneic hematopoietic stem cell transplantation (auto/allo-HSCT). The ARHGAP9high patients undergoing auto/allo-HSCT also had a signifcantly better prognosis with respect to OS and EFS than those receiving only chemotherapy. -

Supplementary Information – Postema Et Al., the Genetics of Situs Inversus Totalis Without Primary Ciliary Dyskinesia
1 Supplementary information – Postema et al., The genetics of situs inversus totalis without primary ciliary dyskinesia Table of Contents: Supplementary Methods 2 Supplementary Results 5 Supplementary References 6 Supplementary Tables and Figures Table S1. Subject characteristics 9 Table S2. Inbreeding coefficients per subject 10 Figure S1. Multidimensional scaling to capture overall genomic diversity 11 among the 30 study samples Table S3. Significantly enriched gene-sets under a recessive mutation model 12 Table S4. Broader list of candidate genes, and the sources that led to their 13 inclusion Table S5. Potential recessive and X-linked mutations in the unsolved cases 15 Table S6. Potential mutations in the unsolved cases, dominant model 22 2 1.0 Supplementary Methods 1.1 Participants Fifteen people with radiologically documented SIT, including nine without PCD and six with Kartagener syndrome, and 15 healthy controls matched for age, sex, education and handedness, were recruited from Ghent University Hospital and Middelheim Hospital Antwerp. Details about the recruitment and selection procedure have been described elsewhere (1). Briefly, among the 15 people with radiologically documented SIT, those who had symptoms reminiscent of PCD, or who were formally diagnosed with PCD according to their medical record, were categorized as having Kartagener syndrome. Those who had no reported symptoms or formal diagnosis of PCD were assigned to the non-PCD SIT group. Handedness was assessed using the Edinburgh Handedness Inventory (EHI) (2). Tables 1 and S1 give overviews of the participants and their characteristics. Note that one non-PCD SIT subject reported being forced to switch from left- to right-handedness in childhood, in which case five out of nine of the non-PCD SIT cases are naturally left-handed (Table 1, Table S1). -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -
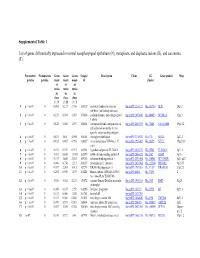
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146.