A Genome-Wide Association Study Lystra P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Breakout Room 5 Poster Presentation Event
Akshara Chandrabalan 2020 NSERC USRA Bioresource Engineering, Supervisor Dr. Prasher 2020 Undergraduate Student Research Awards Predicting Biochar Sorption Capacities Using VIEW THE POSTERS Artificial Neural Networks Poster Presentation Event Breakout Room 5 Daniel Moses 2020 NSERC USRA (ZOOM) SEPTEMBER 17, 2020 @ 4pm Parasitology, Supervisor Dr. Salavati In search of the cryptic motif VI on trypanosomatid RNA editing ligases Richard Boivin 2020 NSERC USRA Antoine Gaudreau 2020 NSERC USRA 1 Natural Resource Sciences, Supervisor Dr. Humphries Bioresource Engineering, Supervisor Dr. Akbarzadeh Muriel Wong Min 2020 AES Brown Martlet USRA Evaluating regional patterns of traditional wildlife 3D Printed Agricultural Wastes for Advanced Food Science & Ag. Chemistry, Supervisor Dr. Karboune harvest in northern Quebec Biocomposites A Database Platform for The Selection of Appropriate Fat Substitutes as "Natural" Katerina Lazaris 2020 NSERC USRA Rowena Groeneveld 2020 NSERC USRA Food Ingredients Animal Science, Supervisor Dr. Bordignon Parasitology, Supervisor Dr. Beech Review: Methods to Improve Oocyte Competence and Modelling of a ligand gated ion channel from embryo Development in Prepubertal Animals to Caenorhabditis elegans experimentally shown to bind Liana Fortin-Hamel 2020 NSERC USRA Accerlerate genetic Gain 6 dopamine Parasitology, Supervisor Dr. Scott 2 Trisha Sackey 2020 NSERC USRA Effects of maternal nematode infection on spatial Food Science & Ag.Chemistry, Supervisor Dr. George Alex Zvezdin 2020 NSERC USRA learning and memory of young mouse pups Natural Resource Sciences, Supervisor Dr. Head Genotypic and Phenotypic Profiling of Staphylo- coccus aureus strains isolated from Canadian Impacts of Agricultural Practices on Yellow Perch Xavier Godin 2020 NSERC USRA Dairy Cattle for Antimicrobial Resistance Spawning Sites in Lac-Saint Pierre Québec Plant Science, Supervisor Dr. -

Investigating Developmental and Functional Deficits in Neurodegenerative
UNIVERSITY OF CALIFORNIA, IRVINE Investigating developmental and functional deficits in neurodegenerative disease using transcriptomic analyses DISSERTATION submitted in partial satisfaction of the requirements for the degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences by Ryan Gar-Lok Lim Dissertation Committee: Professor Leslie M. Thompson, Chair Assistant Professor Dritan Agalliu Professor Peter Donovan Professor Suzanne Sandmeyer 2016 Introduction, Figure 1.1 © 2014 Macmillan Publishers Limited. Appendix 1 © 2016 Elsevier Ltd. All other materials © 2016 Ryan Gar-Lok Lim DEDICATION This dissertation is dedicated to my parents, sister, and my wife. I love you all very much and could not have accomplished any of this without your love and support. Please take the time to reflect back on all of the moments we’ve shared, and know, that it is because of those moments I have been able to succeed. This accomplishment is as much yours as it is mine. ii TABLE OF CONTENTS Page LIST OF FIGURES vi LIST OF TABLES ix ACKNOWLEDGMENTS x CURRICULUM VITAE xiii ABSTRACT OF THE DISSERTATION xv Introduction Huntington’s disease, the neurovascular unit and the blood-brain barrier 1 1.1 Huntington’s Disease 1.2 HTT structure and function 1.2.1 Normal HTT function and possible loss-of-function contributions to HD 1.3 mHTT pathogenesis 1.3.1 The dominant pathological features of mHTT - a gain-of- toxic function? 1.3.2 Cellular pathologies and non-neuronal contributions to HD 1.4 The neurovascular unit and the blood-brain barrier 1.4.1 Structure and function -

Molecular Analyses of Malignant Pleural Mesothelioma
Molecular Analyses of Malignant Pleural Mesothelioma Shir Kiong Lo National Heart and Lung Institute Imperial College Dovehouse Street London SW3 6LY A thesis submitted for MD (Res) Faculty of Medicine, Imperial College London 2016 1 Abstract Malignant pleural mesothelioma (MPM) is an aggressive cancer that is strongly associated with asbestos exposure. Majority of patients with MPM present with advanced disease and the treatment paradigm mainly involves palliative chemotherapy and best supportive care. The current chemotherapy options are limited and ineffective hence there is an urgent need to improve patient outcomes. This requires better understanding of the genetic alterations driving MPM to improve diagnostic, prognostic and therapeutic strategies. This research aims to gain further insights in the pathogenesis of MPM by exploring the tumour transcriptional and mutational profiles. We compared gene expression profiles of 25 MPM tumours and 5 non-malignant pleura. This revealed differentially expressed genes involved in cell migration, invasion, cell cycle and the immune system that contribute to the malignant phenotype of MPM. We then constructed MPM-associated co-expression networks using weighted gene correlation network analysis to identify clusters of highly correlated genes. These identified three distinct molecular subtypes of MPM associated with genes involved in WNT and TGF-ß signalling pathways. Our results also revealed genes involved in cell cycle control especially the mitotic phase correlated significantly with poor prognosis. Through exome analysis of seven paired tumour/blood and 29 tumour samples, we identified frequent mutations in BAP1 and NF2. Additionally, the mutational profile of MPM is enriched with genes encoding FAK, MAPK and WNT signalling pathways. -
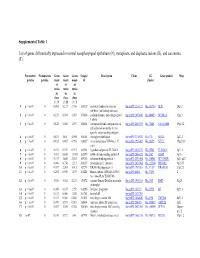
Supplemental Table 1 List of Genes Differentially Expressed In
Supplemental Table 1 List of genes differentially expressed in normal nasopharyngeal epithelium (N), metaplastic and displastic lesions (R), and carcinoma (T). Parametric Permutation Geom Geom Geom Unique Description Clone UG Gene symbol Map p-value p-value mean mean mean id cluster of of of ratios ratios ratios in in in class class class 1 : N 2 : R 3 : T 1 p < 1e-07 0 0.061 0.123 2.708 169329 secretory leukocyte protease IncytePD:2510171 Hs.251754 SLPI 20q12 inhibitor (antileukoproteinase) 2 p < 1e-07 0 0.125 0.394 1.863 163628 sodium channel, nonvoltage-gated IncytePD:1453049 Hs.446415 SCNN1A 12p13 1 alpha 3 p < 1e-07 0 0.122 0.046 1.497 160401 carcinoembryonic antigen-related IncytePD:2060355 Hs.73848 CEACAM6 19q13.2 cell adhesion molecule 6 (non- specific cross reacting antigen) 4 p < 1e-07 0 0.675 1.64 5.594 165101 monoglyceride lipase IncytePD:2174920 Hs.6721 MGLL 3q21.3 5 p < 1e-07 0 0.182 0.487 0.998 166827 nei endonuclease VIII-like 1 (E. IncytePD:1926409 Hs.28355 NEIL1 15q22.33 coli) 6 p < 1e-07 0 0.194 0.339 0.915 162931 hypothetical protein FLJ22418 IncytePD:2816379 Hs.36563 FLJ22418 1p11.1 7 p < 1e-07 0 1.313 0.645 13.593 162399 S100 calcium binding protein P IncytePD:2060823 Hs.2962 S100P 4p16 8 p < 1e-07 0 0.157 1.445 2.563 169315 selenium binding protein 1 IncytePD:2591494 Hs.334841 SELENBP1 1q21-q22 9 p < 1e-07 0 0.046 0.738 1.213 160115 prominin-like 1 (mouse) IncytePD:2070568 Hs.112360 PROML1 4p15.33 10 p < 1e-07 0 0.787 2.264 3.013 167294 HRAS-like suppressor 3 IncytePD:1969263 Hs.37189 HRASLS3 11q12.3 11 p < 1e-07 0 0.292 0.539 1.493 168221 Homo sapiens cDNA FLJ13510 IncytePD:64451 Hs.37896 2 fis, clone PLACE1005146. -

The Conserved ASTN2/BRINP1 Locus at 9Q33.1–33.2 Is Associated With
www.nature.com/scientificreports OPEN The conserved ASTN2/BRINP1 locus at 9q33.1–33.2 is associated with major psychiatric disorders in a large pedigree from Southern Spain Josep Pol‑Fuster1,2, Francesca Cañellas2,3, Laura Ruiz‑Guerra2, Aina Medina‑Dols2, Bàrbara Bisbal‑Carrió1,2, Bernat Ortega‑Vila2,4, Jaume Llinàs1, Jessica Hernandez‑Rodriguez4, Jerònia Lladó1,2, Gabriel Olmos1,2, Konstantin Strauch5,6,7, Damià Heine‑Suñer4, Cristòfol Vives‑Bauzà1,2,8* & Antònia Flaquer5,6,7,8 We investigated the genetic causes of major mental disorders (MMDs) including schizophrenia, bipolar disorder I, major depressive disorder and attention defcit hyperactive disorder, in a large family pedigree from Alpujarras, South of Spain, a region with high prevalence of psychotic disorders. We applied a systematic genomic approach based on karyotyping (n = 4), genotyping by genome‑ wide SNP array (n = 34) and whole‑genome sequencing (n = 12). We performed genome‑wide linkage analysis, family‑based association analysis and polygenic risk score estimates. Signifcant linkage was obtained at chromosome 9 (9q33.1–33.2, LOD score = 4.11), a suggestive region that contains fve candidate genes ASTN2, BRINP1, C5, TLR4 and TRIM32, previously associated with MMDs. Comprehensive analysis associated the MMD phenotype with genes of the immune system with dual brain functions. Moreover, the psychotic phenotype was enriched for genes involved in synapsis. These results should be considered once studying the genetics of psychiatric disorders in other families, especially the ones from the same region, since founder efects may be related to the high prevalence. Psychiatric disorders aggregate in families and their predisposition involve a complex, polygenic and pleiotropic genetic architecture1–3. -

Agricultural University of Athens
ΓΕΩΠΟΝΙΚΟ ΠΑΝΕΠΙΣΤΗΜΙΟ ΑΘΗΝΩΝ ΣΧΟΛΗ ΕΠΙΣΤΗΜΩΝ ΤΩΝ ΖΩΩΝ ΤΜΗΜΑ ΕΠΙΣΤΗΜΗΣ ΖΩΙΚΗΣ ΠΑΡΑΓΩΓΗΣ ΕΡΓΑΣΤΗΡΙΟ ΓΕΝΙΚΗΣ ΚΑΙ ΕΙΔΙΚΗΣ ΖΩΟΤΕΧΝΙΑΣ ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ ΑΘΗΝΑ 2020 ΔΙΔΑΚΤΟΡΙΚΗ ΔΙΑΤΡΙΒΗ Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Genome-wide association analysis and gene network analysis for (re)production traits in commercial broilers ΕΙΡΗΝΗ Κ. ΤΑΡΣΑΝΗ ΕΠΙΒΛΕΠΩΝ ΚΑΘΗΓΗΤΗΣ: ΑΝΤΩΝΙΟΣ ΚΟΜΙΝΑΚΗΣ Τριμελής Επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Επταμελής εξεταστική επιτροπή: Aντώνιος Κομινάκης (Αν. Καθ. ΓΠΑ) Ανδρέας Κράνης (Eρευν. B, Παν. Εδιμβούργου) Αριάδνη Χάγερ (Επ. Καθ. ΓΠΑ) Πηνελόπη Μπεμπέλη (Καθ. ΓΠΑ) Δημήτριος Βλαχάκης (Επ. Καθ. ΓΠΑ) Ευάγγελος Ζωίδης (Επ.Καθ. ΓΠΑ) Γεώργιος Θεοδώρου (Επ.Καθ. ΓΠΑ) 2 Εντοπισμός γονιδιωματικών περιοχών και δικτύων γονιδίων που επηρεάζουν παραγωγικές και αναπαραγωγικές ιδιότητες σε πληθυσμούς κρεοπαραγωγικών ορνιθίων Περίληψη Σκοπός της παρούσας διδακτορικής διατριβής ήταν ο εντοπισμός γενετικών δεικτών και υποψηφίων γονιδίων που εμπλέκονται στο γενετικό έλεγχο δύο τυπικών πολυγονιδιακών ιδιοτήτων σε κρεοπαραγωγικά ορνίθια. Μία ιδιότητα σχετίζεται με την ανάπτυξη (σωματικό βάρος στις 35 ημέρες, ΣΒ) και η άλλη με την αναπαραγωγική -

The Changing Chromatome As a Driver of Disease: a Panoramic View from Different Methodologies
The changing chromatome as a driver of disease: A panoramic view from different methodologies Isabel Espejo1, Luciano Di Croce,1,2,3 and Sergi Aranda1 1. Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology, Dr. Aiguader 88, Barcelona 08003, Spain 2. Universitat Pompeu Fabra (UPF), Barcelona, Spain 3. ICREA, Pg. Lluis Companys 23, Barcelona 08010, Spain *Corresponding authors: Luciano Di Croce ([email protected]) Sergi Aranda ([email protected]) 1 GRAPHICAL ABSTRACT Chromatin-bound proteins regulate gene expression, replicate and repair DNA, and transmit epigenetic information. Several human diseases are highly influenced by alterations in the chromatin- bound proteome. Thus, biochemical approaches for the systematic characterization of the chromatome could contribute to identifying new regulators of cellular functionality, including those that are relevant to human disorders. 2 SUMMARY Chromatin-bound proteins underlie several fundamental cellular functions, such as control of gene expression and the faithful transmission of genetic and epigenetic information. Components of the chromatin proteome (the “chromatome”) are essential in human life, and mutations in chromatin-bound proteins are frequently drivers of human diseases, such as cancer. Proteomic characterization of chromatin and de novo identification of chromatin interactors could thus reveal important and perhaps unexpected players implicated in human physiology and disease. Recently, intensive research efforts have focused on developing strategies to characterize the chromatome composition. In this review, we provide an overview of the dynamic composition of the chromatome, highlight the importance of its alterations as a driving force in human disease (and particularly in cancer), and discuss the different approaches to systematically characterize the chromatin-bound proteome in a global manner. -

Elucidation of Dna Damage Response Pathways And
ELUCIDATION OF DNA DAMAGE RESPONSE PATHWAYS AND FUNCTIONAL EFFICIENCY OF DNA REPAIR GENES ASSOCIATED WITH REPROGRAMMED OSTEOSARCOMA CELL LINES By CHOONG PEI FENG A thesis submitted to the Department of Pre-Clinical Science, Faculty of Medicine and Health Sciences, Universiti Tunku Abdul Rahman, in partial fulfillment of the requirements for the degree of Doctor of Philosophy of Medical Sciences May 2018 ABSTRACT ELUCIDATION OF DNA DAMAGE RESPONSE PATHWAYS AND FUNCTIONAL EFFICIENCY OF DNA REPAIR GENES ASSOCIATED WITH REPROGRAMMED OSTEOSARCOMA CELL LINES Choong Pei Feng Osteosarcoma (OS) is a prevalent cancer of the bone happening mostly in children and adolescence. Alterations and mutations to genes associated with proliferation and differentiation increased the risk of OS tumourigenicity. Reprogramming of OS cell lines to a primitive phase could be used to understand the pathogenesis of OS. Furthermore, the DNA damage response (DDR) of reprogrammed cancer cells has not been well established. By using retroviral OSKM, OS cell lines were reprogrammed to pluripotency. Colonies from iG-292 and iSaos-2 showed ESC-like morphology, expressing pluripotency markers, formed embryoid body-like spheres and expressed markers from three germ layers as well as showing ability to differentiate into adipocytes and osteocytes. However, in vivo study showed teratoma formation only in reprogrammed G-292, iG-292. In the second part of this study, hierarchical clustering analysis from global gene expression profile of parental and reprogrammed OS demonstrated distinctive separation of two population. Differentially expressed genes (DEGs) ii were grouped into DNA repair, cell cycle and apoptosis pathways. Our data showed that iG-292 displayed more DEGs than iSaos-2 in all pathways. -

Autocrine IFN Signaling Inducing Profibrotic Fibroblast Responses By
Downloaded from http://www.jimmunol.org/ by guest on September 23, 2021 Inducing is online at: average * The Journal of Immunology , 11 of which you can access for free at: 2013; 191:2956-2966; Prepublished online 16 from submission to initial decision 4 weeks from acceptance to publication August 2013; doi: 10.4049/jimmunol.1300376 http://www.jimmunol.org/content/191/6/2956 A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Autocrine IFN Signaling Feng Fang, Kohtaro Ooka, Xiaoyong Sun, Ruchi Shah, Swati Bhattacharyya, Jun Wei and John Varga J Immunol cites 49 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2013/08/20/jimmunol.130037 6.DC1 This article http://www.jimmunol.org/content/191/6/2956.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 23, 2021. The Journal of Immunology A Synthetic TLR3 Ligand Mitigates Profibrotic Fibroblast Responses by Inducing Autocrine IFN Signaling Feng Fang,* Kohtaro Ooka,* Xiaoyong Sun,† Ruchi Shah,* Swati Bhattacharyya,* Jun Wei,* and John Varga* Activation of TLR3 by exogenous microbial ligands or endogenous injury-associated ligands leads to production of type I IFN. -

Transcriptome Profiling Reveals the Complexity of Pirfenidone Effects in IPF
ERJ Express. Published on August 30, 2018 as doi: 10.1183/13993003.00564-2018 Early View Original article Transcriptome profiling reveals the complexity of pirfenidone effects in IPF Grazyna Kwapiszewska, Anna Gungl, Jochen Wilhelm, Leigh M. Marsh, Helene Thekkekara Puthenparampil, Katharina Sinn, Miroslava Didiasova, Walter Klepetko, Djuro Kosanovic, Ralph T. Schermuly, Lukasz Wujak, Benjamin Weiss, Liliana Schaefer, Marc Schneider, Michael Kreuter, Andrea Olschewski, Werner Seeger, Horst Olschewski, Malgorzata Wygrecka Please cite this article as: Kwapiszewska G, Gungl A, Wilhelm J, et al. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF. Eur Respir J 2018; in press (https://doi.org/10.1183/13993003.00564-2018). This manuscript has recently been accepted for publication in the European Respiratory Journal. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJ online. Copyright ©ERS 2018 Copyright 2018 by the European Respiratory Society. Transcriptome profiling reveals the complexity of pirfenidone effects in IPF Grazyna Kwapiszewska1,2, Anna Gungl2, Jochen Wilhelm3†, Leigh M. Marsh1, Helene Thekkekara Puthenparampil1, Katharina Sinn4, Miroslava Didiasova5, Walter Klepetko4, Djuro Kosanovic3, Ralph T. Schermuly3†, Lukasz Wujak5, Benjamin Weiss6, Liliana Schaefer7, Marc Schneider8†, Michael Kreuter8†, Andrea Olschewski1, -

The Roles of Complement C4A and C4B Genetic Diversity and HLA DRB1 Variants on Disease Associations with Juvenile Dermatomyositis and Systemic Lupus Erythematosus
The Roles of Complement C4A and C4B Genetic Diversity and HLA DRB1 Variants on Disease Associations with Juvenile Dermatomyositis and Systemic Lupus Erythematosus DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Katherine E. Lintner, B.S. Graduate Program in Molecular, Cellular and Developmental Biology The Ohio State University 2016 Dissertation Committee: Carlos Alvarez, Ph.D. Heithem El-Hodiri, Ph.D. Wael Jarjour, M.D. Yusen Liu, Ph.D. Chack-Yung Yu, D.Phil., Advisor Copyright by Katherine E. Lintner 2016 Abstract Complement C4 is an immune protein with a wide range of effector functions, including disposal of apoptotic materials, clearance of immune complexes, and activation of the classical complement pathway resulting in cytolysis of microbes. Homozygous deficiencies of C4 or early complement components (C1q, C1r, or C1s), albeit rare, are strongly associated with the autoimmune disease systemic lupus erythematosus (SLE). Much more common than a complete genetic deficiency is “low” gene copy number (GCN) of C4, which varies among human genomes from two to eight copies. Low GCN of C4, specifically of the isotype C4A, is associated with SLE disease risk. However, it is known that C4A-deficient haplotypes in European Americans are in strong linkage disequilibrium (LD) with HLA allele DRB1*0301 on chromosome 6, which has been associated with other autoimmune diseases, including juvenile dermatomyositis (JDM). It remains a puzzle whether C4A deficiency, DRB1*0301, or both are responsible for the primary disease association because of the strong LD exhibited between the two genetic variants. -

Assessing Conservation of Alternative Splicing with Evolutionary Splicing Graphs
Assessing conservation of alternative splicing with evolutionary splicing graphs Supplemental Material 1 2 3 Diego Javier Zea , Sofya Laskina , Alexis Baudin , 1,2∗ 1∗ Hugues Richard and Elodie Laine 1 Sorbonne Universit´e, CNRS, IBPS, Laboratoire de Biologie Computationnelle et Quantitative (LCQB), 75005 Paris, France. 2 Bioinformatics Unit (MF1), Department for Methods development and Research Infrastructure, Robert Koch Institute, 13353 Berlin, Germany. 3 Sorbonne Universit´e, CNRS, LIP6, F- 75005 Paris, France. ∗ corresponding authors: [email protected], [email protected] 1 Supplemental Methods Datasets AS-dedicated gene set We collected a set of 50 genes, representing 16 families, where AS produces functionally distinct protein isoforms (Supplemental Table S1). Within each family, the biochemical activities of several isoforms have been characterised. The set contains single-domain genes as well as multi- domain ones, ranging from less than 200 to 2000 residues. It includes some kinases, some receptors, some RNA-binding proteins, a transcription factor, and a significant portion of proteins involved in the formation of the cell cytoskeleton, in muscle contraction and in membrane trafficking. Human proteome We considered the ensemble of 19,976 protein coding genes comprised in the human genome and their one-to-one orthologs from gorilla, macaque, mouse, rat, boar, cow, opossum, platypus, frog, zebrafish and nematode. We downloaded the corresponding gene annotations from Ensembl1 release 98 (September 2019). The download was successful for 18 241 genes. Among those, 14 genes did not have any good quality transcripts (see below for the criteria) and 1 gene displayed an error in the gene tree, leading to a total of 18 226 valid genes.