ORGANIZATION of BIOMOLECULES in a 3-DIMENSIONAL SPACE by Sameer Sajja a Thesis Submitted to the Faculty of the University Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Effects of Diatom Pore-Size on the Structures and Extensibilities of Single Mucilage Molecules
Edinburgh Research Explorer The effects of diatom pore-size on the structures and extensibilities of single mucilage molecules Citation for published version: Sanka, I, Suyono, E & Alam, P 2017, 'The effects of diatom pore-size on the structures and extensibilities of single mucilage molecules', Carbohydrate Research, vol. 448, pp. 35-42. https://doi.org/10.1016/j.carres.2017.05.014 Digital Object Identifier (DOI): 10.1016/j.carres.2017.05.014 Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: Carbohydrate Research General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 06. Oct. 2021 Carbohydrate Research 448 (2017) 35e42 Contents lists available at ScienceDirect Carbohydrate Research journal homepage: www.elsevier.com/locate/carres The effects of diatom pore-size on the structures and extensibilities of single mucilage molecules * Immanuel Sanka a, b, Eko Agus -

Mai Muuttunut Pilit Muut Aidi Mini
MAIMUUTTUNUT US009963689B2 PILIT MUUT AIDI MINI (12 ) United States Patent ( 10 ) Patent No. : US 9 ,963 , 689 B2 Doudna et al. ( 45) Date of Patent: May 8 , 2018 ( 54 ) CASI CRYSTALS AND METHODS OF USE FOREIGN PATENT DOCUMENTS THEREOF WO WO 2013 / 126794 AL 8 / 2013 ( 71 ) Applicant: The Regents of the University of wo WO 2013 / 142578 A19 / 2013 California , Oakland , CA (US ) WO WO 2013 / 176772 A1 11/ 2013 ( 72 ) Inventors : Jennifer A . Doudna , Oakland , CA OTHER PUBLICATIONS (US ) ; Samuel H . Sternberg , Oakland , McPherson , A . Current Approaches to Macromolecular Crystalli CA (US ) ; Martin Jinek , Oakland , CA zation . European Journal of Biochemistry . 1990 . vol . 189 , pp . (US ) ; Fuguo Jiang , Oakland , CA (US ); 1 - 23 . * Emine Kaya , Oakland , CA (US ) ; Kundrot, C . E . Which Strategy for a Protein Crystallization Project ? Cellular Molecular Life Science . 2004 . vol . 61, pp . 525 - 536 . * David W . Taylor, Jr. , Oakland , CA Benevenuti et al. , Crystallization of Soluble Proteins in Vapor (US ) Diffusion for X - ray Crystallography , Nature Protocols , published on - line Jun . 28 , 2007 , 2 ( 7 ) : 1633 - 1651. * ( 73 ) Assignee : THE REGENTS OF THE Cudney R . Protein Crystallization and Dumb Luck . The Rigaku UNIVERSITY OF CALIFORNIA , Journal. 1999 . vol. 16 , No . 1 , pp . 1 - 7 . * Drenth , “ Principles of Protein X - Ray Crystallography ” , 2nd Edi Oakland , CA (US ) tion , 1999 Springer - Verlag New York Inc ., Chapter 1 , p . 1 - 21. * Moon et al . , “ A synergistic approach to protein crystallization : ( * ) Notice : Subject to any disclaimer , the term of this Combination of a fixed -arm carrier with surface entropy reduction ” , patent is extended or adjusted under 35 Protein Science , 2010 , 19 : 901 -913 . -

Pelvic Fin Flexibility in Tree Climbing Fish
G Model ZOOL-25524; No. of Pages 7 ARTICLE IN PRESS Zoology xxx (2016) xxx–xxx Contents lists available at ScienceDirect Zoology journal homepage: www.elsevier.com/locate/zool The significance of pelvic fin flexibility for tree climbing fish a b a,b c Adhityo Wicaksono , Saifullah Hidayat , Yudithia Damayanti , Desmond Soo Mun Jin , a b,∗ a,∗ Erly Sintya , Bambang Retnoaji , Parvez Alam a Laboratory of Paper Coating and Converting, Centre for Functional Materials, Abo Akademi University, Porthaninkatu 3, 20500 Turku, Finland b Laboratory of Animal Embryology, Faculty of Biology, Universitas Gadjah Mada, Yogyakarta, Indonesia c Rapid Gain Global Corporation, Singapore a r t i c l e i n f o a b s t r a c t Article history: In this article, we compare the characteristics of biomechanical attachment exhibited by two morpholog- Received 1 March 2016 ically different mudskipper species, Boleophthalmus boddarti (with fused pelvic fins) and Periophthalmus Received in revised form 14 April 2016 variabilis (with unfused pelvic fins). P. variabilis is a tree and rock climber while B. boddarti dwells in the Accepted 17 June 2016 muddy shallows and is unable to climb. Our aim in this article is to determine whether it is predominantly Available online xxx chemical or morphological properties of the pelvic fins from each species that may allow P. variabilis to climb trees whilst preventing B. boddarti from doing the same. To fulfil our objective we perform friction Keywords: and suction resistance tests, Fourier transform infrared spectroscopy of the mucosal secretions under Mudskipper the fins, direct geometrical measurements and finite element modelling. -

Advances in Computational Fluid Mechanics in Cellular Flow Manipulation: a Review
applied sciences Review Advances in Computational Fluid Mechanics in Cellular Flow Manipulation: A Review Masoud Arabghahestani 1,* , Sadegh Poozesh 2 and Nelson K. Akafuah 1 1 Institute of Research for Technology Development (IR4TD), University of Kentucky, Lexington, KY 40506, USA; [email protected] 2 Mechanical Engineering Department, Tuskegee University, Tuskegee, AL 36088, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-(859)-898-7700 Received: 28 August 2019; Accepted: 25 September 2019; Published: 27 September 2019 Abstract: Recently, remarkable developments have taken place, leading to significant improvements in microfluidic methods to capture subtle biological effects down to single cells. As microfluidic devices are getting sophisticated, design optimization through experimentations is becoming more challenging. As a result, numerical simulations have contributed to this trend by offering a better understanding of cellular microenvironments hydrodynamics and optimizing the functionality of the current/emerging designs. The need for new marketable designs with advantageous hydrodynamics invokes easier access to efficient as well as time-conservative numerical simulations to provide screening over cellular microenvironments, and to emulate physiological conditions with high accuracy. Therefore, an excerpt overview on how each numerical methodology and associated handling software works, and how they differ in handling underlying hydrodynamic of lab-on-chip microfluidic is crucial. These numerical means rely on molecular and continuum levels of numerical simulations. The current review aims to serve as a guideline for researchers in this area by presenting a comprehensive characterization of various relevant simulation techniques. Keywords: computational fluid mechanics; microfluidic devices; cellular flow; numerical simulations; molecular and continuum levels 1. Introduction 1.1. -

Amino Acid Biomaterials
Edinburgh Research Explorer Mechanical properties of TEMPO-oxidised bacterial cellulose- amino acid biomaterials Citation for published version: Pahlevan, M, Toivakka, M & Alam, P 2018, 'Mechanical properties of TEMPO-oxidised bacterial cellulose- amino acid biomaterials', European Polymer Journal, vol. 101, pp. 29-36. https://doi.org/10.1016/j.eurpolymj.2018.02.013 Digital Object Identifier (DOI): 10.1016/j.eurpolymj.2018.02.013 Link: Link to publication record in Edinburgh Research Explorer Document Version: Peer reviewed version Published In: European Polymer Journal General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 30. Sep. 2021 Mechanical properties of TEMPO-oxidised bacterial cellulose- amino acid biomaterials Mahdi Pahlevan1, Martti Toivakka1, Parvez Alam1,2 1 Laboratory of Paper Coating and Converting, Centre for Functional Materials, Abo Akademi University, Turku, Finland 2 School of Engineering, Institute for Materials and Processes, University of Edinburgh, United Kingdom *Corresponding Author: Parvez Alam, School of Engineering, Institute for Materials and Processes, University of Edinburgh, United Kingdom. Email: [email protected] and [email protected] Keywords: Nanocellulose, TEMPO-Oxidation, Silk Bioglue, Amino Acid Abstract Amino acid functionalised bacterial cellulose is a non-toxic biocompatible material, which can be further modified with active groups and nanoparticles for various biomedical applications. -

WHAT INFLUENCE WOULD a CLOUD BASED SEMANTIC LABORATORY NOTEBOOK HAVE on the DIGITISATION and MANAGEMENT of SCIENTIFIC RESEARCH? by Samantha Kanza
UNIVERSITY OF SOUTHAMPTON Faculty of Physical Sciences and Engineering School of Electronics and Computer Science What Influence would a Cloud Based Semantic Laboratory Notebook have on the Digitisation and Management of Scientific Research? by Samantha Kanza Thesis for the degree of Doctor of Philosophy 25th April 2018 UNIVERSITY OF SOUTHAMPTON ABSTRACT FACULTY OF PHYSICAL SCIENCES AND ENGINEERING SCHOOL OF ELECTRONICS AND COMPUTER SCIENCE Doctor of Philosophy WHAT INFLUENCE WOULD A CLOUD BASED SEMANTIC LABORATORY NOTEBOOK HAVE ON THE DIGITISATION AND MANAGEMENT OF SCIENTIFIC RESEARCH? by Samantha Kanza Electronic laboratory notebooks (ELNs) have been studied by the chemistry research community over the last two decades as a step towards a paper-free laboratory; sim- ilar work has also taken place in other laboratory science domains. However, despite the many available ELN platforms, their uptake in both the academic and commercial worlds remains limited. This thesis describes an investigation into the current ELN landscape, and its relationship with the requirements of laboratory scientists. Market and literature research was conducted around available ELN offerings to characterise their commonly incorporated features. Previous studies of laboratory scientists examined note-taking and record-keeping behaviours in laboratory environments; to complement and extend this, a series of user studies were conducted as part of this thesis, drawing upon the techniques of user-centred design, ethnography, and collaboration with domain experts. These user studies, combined with the characterisation of existing ELN features, in- formed the requirements and design of a proposed ELN environment which aims to bridge the gap between scientists' current practice using paper lab notebooks, and the necessity of publishing their results electronically, at any stage of the experiment life cycle. -
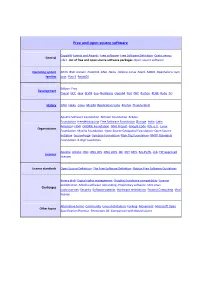
Free and Open Source Software
Free and open source software Copyleft ·Events and Awards ·Free software ·Free Software Definition ·Gratis versus General Libre ·List of free and open source software packages ·Open-source software Operating system AROS ·BSD ·Darwin ·FreeDOS ·GNU ·Haiku ·Inferno ·Linux ·Mach ·MINIX ·OpenSolaris ·Sym families bian ·Plan 9 ·ReactOS Eclipse ·Free Development Pascal ·GCC ·Java ·LLVM ·Lua ·NetBeans ·Open64 ·Perl ·PHP ·Python ·ROSE ·Ruby ·Tcl History GNU ·Haiku ·Linux ·Mozilla (Application Suite ·Firefox ·Thunderbird ) Apache Software Foundation ·Blender Foundation ·Eclipse Foundation ·freedesktop.org ·Free Software Foundation (Europe ·India ·Latin America ) ·FSMI ·GNOME Foundation ·GNU Project ·Google Code ·KDE e.V. ·Linux Organizations Foundation ·Mozilla Foundation ·Open Source Geospatial Foundation ·Open Source Initiative ·SourceForge ·Symbian Foundation ·Xiph.Org Foundation ·XMPP Standards Foundation ·X.Org Foundation Apache ·Artistic ·BSD ·GNU GPL ·GNU LGPL ·ISC ·MIT ·MPL ·Ms-PL/RL ·zlib ·FSF approved Licences licenses License standards Open Source Definition ·The Free Software Definition ·Debian Free Software Guidelines Binary blob ·Digital rights management ·Graphics hardware compatibility ·License proliferation ·Mozilla software rebranding ·Proprietary software ·SCO-Linux Challenges controversies ·Security ·Software patents ·Hardware restrictions ·Trusted Computing ·Viral license Alternative terms ·Community ·Linux distribution ·Forking ·Movement ·Microsoft Open Other topics Specification Promise ·Revolution OS ·Comparison with closed -
S.A. Raja Pharmacy College Vadakkangulam-627 116
S.A. RAJA PHARMACY COLLEGE VADAKKANGULAM-627 116 MEDICINAL CHEMISTRY -III VI SEMESTER B. PHARM PRACTICAL MANUAL CONTENT S.No Experiment Name Page No. 1. Synthesis of Sulphanilamide 01 2. Synthesis of 7- Hydroxy -4- methyl coumarin 03 3. Synthesis of Chlorbutanol 05 4. Synthesis of Tolbutamide 07 5. Synthesis of Hexamine 09 6. Assay of Isonicotinic acid hydrazide 11 7. Assay of Metronidazole 13 8. Assay of Dapsone 16 9. Assay of Chlorpheniramine Maleate 18 10. Assay of Benzyl Penicillin 20 11. Synthesis of Phenytoin from Benzil by Microwave 23 Irradiation 12. Synthesis of Aspirin Assisted by Microwave Oven 26 13. Drawing structure and Reaction using Chemsketch 28 MEDICINAL CHEMISTRY- III Experiment No: 01 Synthesis of Sulphanilamide Aim: To synthesis and submit sulphanilamide from p-acetamido benzene sulphanilamide and calculate its percentage yield. Principle: Sulphanilamide can be prepared by the reaction of P-acetamido benzene sulphanilamide with Hydrochloric acid or ammonium carbonate. The acetamido groups are easily undergo acid catalysed hydrolysis reaction to form p-amino benzene sulphonamide. Reaction: O HN H2N HCl O S O O S O NH NH2 2 4 Acetamidobenzene sulphonamide p Amino benzene sulphonamide Chemical Required: Resorcinol - 1.2 g Ethyl acetoacetate - 2.4 ml Conc. Sulphuric acid - 7.5 ml Procedure: 1.5 gm of 4- acetamido benzene sulphonamide is treated with a mixture of 1 ml of conc. Sulphuric acid diluted with 2 ml water. This mixture is gently heated under reflux for 1 hour. Then 3ml of water is added and the solution is boiled again, with the addition of a small quantity of activated charcoal. -
Isurvey - Online Questionnaire Generation from the University of Southampton
1/17/2018 iSurvey - Online Questionnaire Generation from the University of Southampton Investigating the use of software for Chemists Survey Study Information Study title: Investigating the use of software for Chemists Survey Researcher name: Samantha Kanza Study reference: iSurvey 16857 Ethics reference: Ergo 17642 – Chemistry Tools Survey Participant Information Please read this information carefully before deciding to take part in this research. If you are happy to participate you will be asked to check the consent form box. What is the research about? This research is for my PhD in Computer Science and Chemistry. I am coming to the end of the first year of my PhD and I am looking to investigate the use of chemistry tools to better understand what type of tools chemists actually use. This is a very simple survey that asks what type of chemist you are, and what types of tools you use, and if applicable which specific tools of that type you use. This PhD is part of the Web Science CDT and is funded by EPSRC. Why have I been chosen? You have been chosen because you work in chemistry. What will happen to me if I take part? This is a short survey that is being conducted to get a better idea of the usage of chemistry tools. Are there any benefits in my taking part? This survey will form part of a body of research aimed to improve the understanding of how chemists use technology to assist their work. Are there any risks involved? There are no risks involved. Will my participation be confidential? Your participation will be confidential. -
Characterisation of a Solvent-Tolerant Haloarchaeal (R)-Selective Transaminase Isolated from a Triassic Period Salt Mine
Characterisation of a solvent-tolerant haloarchaeal (R)-selective transaminase isolated from a Triassic period salt mine Kelly, S., Magill, D., Megaw, J., Skvortsov, T., Allers, T., McGrath, J., Allen, C., Moody, T. S., & Gilmore, B. (2019). Characterisation of a solvent-tolerant haloarchaeal (R)-selective transaminase isolated from a Triassic period salt mine. Applied Microbiology and Biotechnology, 103, 5727. https://doi.org/10.1007/s00253-019- 09806-y Published in: Applied Microbiology and Biotechnology Document Version: Publisher's PDF, also known as Version of record Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal Publisher rights Copyright 2019 the authors. This is an open access article published under a Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. If you discover content in the Research Portal that you believe breaches copyright or violates any law, please contact [email protected]. Download date:01. -

Artificial Biomineralisation of Flax Fibres in the Presence of Amino Acids for Use in Natural Fibre Reinforced Composites
UNIVERSITÀ DEGLI STUDI DI PADOVA DIPARTIMENTO DI INGEGNERIA INDUSTRIALE CORSO DI LAUREA MAGISTRALE IN INGEGNERIA CHIMICA E DEI PROCESSI INDUSTRIALI Tesi di Laurea Magistrale in Ingegneria Chimica e dei Processi Industriali ARTIFICIAL BIOMINERALISATION OF FLAX FIBRES IN THE PRESENCE OF AMINO ACIDS FOR USE IN NATURAL FIBRE REINFORCED COMPOSITES Relatore: Prof. Manuele Dabalà Correlatore: Prof.(adj.) Parvez Alam, Åbo Akademi University (FIN) Laureando: ANDREA CALZAVARA ANNO ACCADEMICO 2013 – 2014 Riassunto Negli ultimi anni la comunità scientifica si è focalizzata sullo sviluppo di criteri per la salvaguardia ambientale che accompagnino la progettazione di un determinato bene. Questa tendenza si traduce anche in innovazione nella scienza e nella tecnologia con l’obiettivo di ridurre le emissioni di gas ad effetto serra e le quantità di sostanze che sono difficilmente riciclabili alla fine del loro ciclo di vita; il tutto, senza però rinunciare alle prestazioni del prodotto. Nel campo dei materiali compositi, i rinforzi fibrosi naturali e le matrici biopolimeriche diventano un logico approccio verso uno sviluppo sostenibile, ma l’elevato costo dei biopolimeri non consente loro un ampio utilizzo. La via più praticabile verso materiali compositi eco-compatibili risulta quindi, per ora, l’utilizzo, ove possibile, di fibre naturali nei materiali compositi rinforzati a fibre. Il riciclo, la biodegradabilità e la combustibilità non sono le uniche ragioni che promuovono lo sviluppo di tecniche innovative riguardanti le fibre rinnovabili. Altri vantaggi -

Characterisation of a Solvent-Tolerant Haloarchaeal (R)-Selective Transaminase Isolated from a Triassic Period Salt Mine
Characterisation of a solvent-tolerant haloarchaeal (R)-selective transaminase isolated from a Triassic period salt mine Kelly, S., Magill, D., Megaw, J., Skvortsov, T., Allers, T., McGrath, J., ... Gilmore, B. (2019). Characterisation of a solvent-tolerant haloarchaeal (R)-selective transaminase isolated from a Triassic period salt mine. Applied Microbiology and Biotechnology. https://doi.org/10.1007/s00253-019-09806-y Published in: Applied Microbiology and Biotechnology Document Version: Publisher's PDF, also known as Version of record Queen's University Belfast - Research Portal: Link to publication record in Queen's University Belfast Research Portal Publisher rights Copyright 2019 the authors. This is an open access article published under a Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. General rights Copyright for the publications made accessible via the Queen's University Belfast Research Portal is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The Research Portal is Queen's institutional repository that provides access to Queen's research output. Every effort has been made to ensure that content in the Research Portal does not infringe any person's rights, or applicable UK laws. If you discover content in the Research Portal that you believe breaches copyright or violates any law, please contact [email protected]. Download date:01. Jun. 2019 Applied Microbiology and Biotechnology https://doi.org/10.1007/s00253-019-09806-y BIOTECHNOLOGICALLY RELEVANT ENZYMES AND PROTEINS Characterisation of a solvent-tolerant haloarchaeal (R)-selective transaminase isolated from a Triassic period salt mine Stephen A.