Bonviva, INN- Ibandronic Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CCA Senior Care Options Formulary
Commonwealth Care Alliance Senior Care Option HMO SNP 2021 List of Covered Drugs Formulary 30 Winter Street • Boston, MA 02108 PLEASE READ: THIS DOCUMENT CONTAINS INFORMATION ABOUT THE DRUGS WE COVER IN THIS PLAN This formulary was updated on 08/01/2021. For more recent information or other questions, please contact Senior Care Options Program (HMO SNP) Member Services, at 1-866-610-2273 or, for TTY users, 711, 8 a.m. – 8 p.m., 7 days a week, or visit www.commonwealthcaresco.org. HPMS Approved Formulary File Submission ID 00021589, Version Number 13 Senior Care Options Program (HMO SNP) 2021 Formulary (List of Covered Drugs) PLEASE READ: THIS DO CUMENT CONTAINS INFORMATION ABOUT THE DRUGS WE COVER IN THIS PLAN HPMS Approved Formulary File Submission ID 00021589, Version Number 13 Note to existing members: This formulary has changed since last year. Please review this document to make sure that it still contains the drugs you take. When this drug list (formulary) refers to “we,” “us”, or “our,” it means Commonwealth Care Alliance. When it refers to “plan” or “our plan,” it means 2021 Senior Care Options Program. This document includes list of the drugs (formulary) for our plan which is current as of 08/01/2021. This formulary document applies to all SCO members. For an updated formulary, please contact us. Our contact information, along with the date we last updated the formulary, appears on the front and back cover pages. You must generally use network pharmacies to use your prescription drug benefit. Benefits, formulary, pharmacy n etwork, and/or copayments/coinsurance may change on January 1, 2022, and from time to time during the year. -

Bisphosphonate Use in Patients with Breast Cancer
Bisphosphonate use in patients with breast cancer Information for patients, relatives and carers For more information, please contact: Medicines Information Email: [email protected] York Hospital Medicines Information Tel: 01904 725960 Page 2 Contents Page Why have I been given this leaflet? ............................... 4 How is the treatment given? .......................................... 4 How long will I need to take the treatment for? .............. 6 Can I take other medicines at the same time? ............... 6 Who will prescribe the medication? ............................... 7 What if I am already on bisphosphonate treatment? ...... 7 Bisphosphonates are not licensed for reducing the risk of breast cancer recurrence. What does this mean? ...... 8 Are there any side effects? ............................................ 9 What is osteonecrosis? ............................................... 10 Are there any signs and symptoms I should look out for during treatment? ........................................................ 11 How can I decrease the risk of developing osteonecrosis of the jaw? ................................................................... 12 What do I need to do before starting treatment? .......... 13 Will I require any monitoring, or blood tests? ............... 14 Should I stop taking the bisphosphonate? ................... 14 Tell us what you think of this leaflet ............................. 15 Teaching, training and research ................................... 15 Patient Advice and Liaison Service (PALS) .................. 15 Page 3 Why have I been given this leaflet? You have been given this leaflet because you are going to be treated with a bisphosphonate medicine (zoledronic acid and/or ibandronic acid). In post-menopausal women, with early breast cancer, these drugs have been shown to reduce the risk of the disease recurring and increase length of life. How is the treatment given? The treatment will usually be provided as an oral tablet of ibandronic acid (one 50mg tablet taken once daily). -
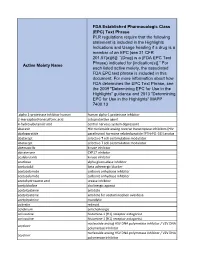
Active Moiety Name FDA Established Pharmacologic Class (EPC) Text
FDA Established Pharmacologic Class (EPC) Text Phrase PLR regulations require that the following statement is included in the Highlights Indications and Usage heading if a drug is a member of an EPC [see 21 CFR 201.57(a)(6)]: “(Drug) is a (FDA EPC Text Phrase) indicated for [indication(s)].” For Active Moiety Name each listed active moiety, the associated FDA EPC text phrase is included in this document. For more information about how FDA determines the EPC Text Phrase, see the 2009 "Determining EPC for Use in the Highlights" guidance and 2013 "Determining EPC for Use in the Highlights" MAPP 7400.13. .alpha. -

Illness Code A
Long Term Illness (LTI) Scheme: Reference Guide as to what is covered in the Health Service Executive for each of the 16 scheduled LTI illnesses. (January 2021) Illness Code C: Cerebral Palsy Antibiotics 1. Doxycycline J01AA02 2. Lymecycline J01AA04 3. Minocycline J01AA08 4. Ampicillin J01CA01 5. Amoxicillin J01CA04 6. Azlocillin J01CA09 7. Benzylpenicillin J01CE01 8. Phenoxymethylpenicillin J01CE02 9. Flucloxacillin J01CF05 10. Amoxicillin & Enzyme Inhibitor J01CR02 11. Cefalexin J01DB01 12. Cefuroxime J01DC02 13. Cefaclor J01DC04 14. Ceftazidime J01DD02 15. Ceftriaxone J01DD04 16. Cefixime J01DD08 17. Cefpodoxime J01DD13 18. Trimethoprim J01EA01 19. Sulfamethoxazole & Trimethoprim J01EE01 20. Erythromycin J01FA01 21. Clarithromycin J01FA09 22. Azithromycin J01FA10 23. Clindamycin J01FF01 24. Gentamicin J01GB03 25. Amikacin J01GB06 26. Ofloxacin J01MA01 27. Ciprofloxacin J01MA02 28. Levofloxacin J01MA12 29. Moxifloxacin J01MA14 30. Vancomycin J01XA01 31. Teicoplanin J01XA02 32. Colistin J01XB01 33. Fusidic Acid J01XC01 34. Nitrofurantoin J01XE01 Muscle relaxants 1. Baclofen M03BX01 2. Tizanidine M03BX02 3. Dantrolene M03CA01 Anticonvulsants (including Benzodiazepines used for this purpose) 1. Phenobarbital N03AA02 2. Primidone N03AA03 page 1 3. Phenytoin N03AB02 4. Ethosuximide N03AD01 5. Clonazepam N03AE01 6. Carbamazepine N03AF01 7. Oxcarbazepine N03AF02 8. Rufinamide N03AF03 9. Eslicarbazepine N03AF04 10. Valproic Acid N03AG01 11. Vigabatrin N03AG04 12. Tiagabine N03AG06 13. Lamotrigine N03AX09 14. Topiramate N03AX11 15. Gabapentin N03AX12 16. Levetiracetam N03AX14 17. Zonisamide N03AX15 18. Pregabalin N03AX16 19. Lacosamide N03AX18 20. Retigabine N03AX21 21. Perampanel N03AX22 22. Diazepam (Rectal) N05BA01 23. Clobazam N05BA09 24. Midazolam N05CD08 25. Acetazolamide S01EC01 Anxiolytics 1. Diazepam N05BA01 2. Chlordiazepoxide N05BA02 3. Lorazepam N05BA06 4. Bromazepam N05BA08 5. Clobazam N05BA09 6. Prazepam N05BA11 7. Alprazolam N05BA12 Hypnotics 8. -
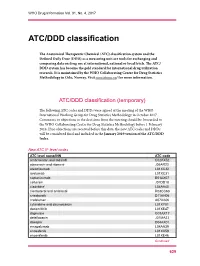
ATC/DDD Classification
WHO Drug Information Vol. 31, No. 4, 2017 ATC/DDD classification The Anatomical Therapeutic Chemical (ATC) classification system and the Defined Daily Dose (DDD) as a measuring unit are tools for exchanging and comparing data on drug use at international, national or local levels. The ATC/ DDD system has become the gold standard for international drug utilization research. It is maintained by the WHO Collaborating Centre for Drug Statistics Methodology in Oslo, Norway. Visit www.whocc.no/ for more information. ATC/DDD classification (temporary) The following ATC codes and DDDs were agreed at the meeting of the WHO International Working Group for Drug Statistics Methodology in October 2017. Comments or objections to the decisions from the meeting should be forwarded to the WHO Collaborating Centre for Drug Statistics Methodology before 1 February 2018. If no objections are received before this date, the new ATC codes and DDDs will be considered final and included in the January 2019 version of the ATC/DDD Index. New ATC 5th level codes ATC level name/INN ATC code ambrisentan and tadalafil C02KX52 atazanavir and ritonavir J05AR23 atezolizumab L01XC32 avelumab L01XC31 caplacizumab B01AX07 cefteram J01DD18 cladribine1 L04AA40 clenbuterol and ambroxol R03CC63 crisaborole D11AH06 crofelemer A07XA06 cytarabine and daunorubicin L01XY01 dacomitinib L01XE47 dapivirine G01AX17 delafloxacin J01MA23 doxepin D04AX01 emapalumab L04AA39 enasidenib L01XX59 encorafenib L01XE46 Continued 629 ATC/DDD classification WHO Drug Information Vol. 31, No. 4, 2017 -

Position Paper American Association of Oral and Maxillofacial Surgeons
saving faces|changing lives® Position Paper American Association of Oral and Maxillofacial Surgeons Medication-Related Osteonecrosis of the Jaw—2014 Update Special Committee on Medication- Introduction Related Osteonecrosis of the Jaws: The Special Committee recommends changing the Salvatore L. Ruggiero, DMD, MD, Clinical nomenclature of bisphosphonate-related osteonecrosis of Professor, Division of Oral and Maxillofacial the jaw (BRONJ). The Special Committee favors the term Surgery, Stony Brook School of Dental Medicine, medication-related osteonecrosis of the jaw (MRONJ). Hofstra North Shore-LIJ School of Medicine, The change is justifed to accommodate the growing New York Center for Orthognathic and Maxillo- number of osteonecrosis cases involving the maxilla and facial Surgery, Lake Success, NY mandible associated with other antiresorptive (denosumab) and antiangiogenic therapies. Thomas B. Dodson, DMD, MPH, Professor and Chair, Associate Dean for Hospital Affairs, MRONJ adversely affects the quality of life, produc- University of Washington School of Dentistry, ing signifcant morbidity. Strategies for management of Department of Oral and Maxillofacial Surgery, patients with, or at risk for, MRONJ were set forth in the Seattle, WA American Association of Oral and Maxillofacial Sur- geons (AAOMS) updated Position Paper on Bisphospho- John Fantasia, DDS, Chief, Division of Oral nate-Related Osteonecrosis of the Jaws and approved by Pathology, Hofstra North Shore-LIJ School of the Board of Trustees in 2009.1 The Position Paper was Medicine , New Hyde Park, NY developed by a Special Committee appointed by the Board Reginald Goodday, Professor, Department of Oral and composed of clinicians with extensive experience in and Maxillofacial Sciences, Dalhousie University, caring for these patients and basic science researchers. -

Recommendations from York and Scarborough Medicines Commissioning Committee September 2017
Recommendations from York and Scarborough Medicines Commissioning Committee September 2017 Drug name Indication Recommendation, rationale and place in therapy RAG status Potential full year cost impact 1 TA463: Cabozantinib for previously Recommended as an option for treating advanced Red No cost impact to CCGs as NHS treated advanced renal cell renal cell carcinoma in adults after VEGF-targeted England commissioned. carcinoma therapy, only if company provides it with discount agreed in the PAS. 2 TA464: Bisphosphonates for Oral bisphosphonates (alendronic acid, ibandronic acid All drugs Low/no cost impact expected. It is not treating osteoporosis and risedronate sodium) are recommended as options listed in the expected that practice will change for treating osteoporosis in adults only if: guidance are substantially as a result of this the person is eligible for risk assessment as already on guidance. defined in NICE's guideline on osteoporosis and formulary as green (oral) the 10‑year probability of osteoporotic fragility or red (IV) for fracture is at least 1%. osteoporosis Intravenous bisphosphonates (ibandronic acid and zoledronic acid) are recommended as options for treating osteoporosis in adults only if: the person is eligible for risk assessment as defined in NICE's guideline on osteoporosis and the 10‑year probability of osteoporotic fragility fracture is at least 10% or the 10‑year probability of osteoporotic fragility fracture is at least 1% and the person has difficulty taking oral bisphosphonates (alendronic acid, ibandronic acid or risedronate sodium) or these drugs are contraindicated or not tolerated. 3 TA465: Olaratumab in combination Olaratumab, in combination with doxorubicin, is Red No cost impact to CCGs as NHS with doxorubicin for treating recommended for use within the Cancer Drugs Fund England commissioned. -

“Wherever the Art of Medicine Is Loved, There Is Also Love of Humanity" Hipócrates ABOUT US
“Wherever the art of Medicine is loved, there is also love of Humanity" Hipócrates ABOUT US CATENA GROUP provides complete and vertical services, we act as a key link for all our clients as well as offering to obtain various benefits in their purchases. Such as simplification of procedures through the know-how of our company, relationships and experience in the different sectors EXPLANATION & PHILOSOPHY that we are specialized in, these are just some of the unique CATENA FROM LATIN, CATENA, /ka'te:.na/ advantages on collaborating with us. Circular object formed by interlaced links. Intangible bond that unites people. Succession of related facts or phenomena. Consequently, we are able to satisfy 100% of our customer‘s needs through our networks of contacts, control, dedication and engagement in every operation. CATENA MEDICAL SERVICES operates We provide all of the services to facilitate Subsequently, our work with more than 778 mainly as a purchasing center (not as a our client’s work with first level laboratories references of generic medicines (including distributor) for Governments and State or from countries such as France, Germany, Food Supplements, Vitamins, Women's Private Companies. Greece, Italy, Spain, etc. We assist them by hygiene products, etc.) and more than 30 handling the negotiations in order to top-level laboratories, one of the key unique We are specialists in providing solutions to achieve their target prices and orders, advantages of working with us is our ability our client’s requirements. simplify document management and to adapt the research of our team, in order provide logistics and legal support if to provide the most interesting products required, etc. -

Brand to Generic Switches
Brand to Generic switches Original Product Strength Pack Size Cost per Change to Pack size Cost per Savings per Notes Annual month month month savings/ pack 15mg £25.83 £1.63 £24.20 £314.60 Actos® tablets 30mg 28 £35.89 Pioglitazone tablets 28 £1.90 £33.99 £441.87 45mg £39.55 £2.62 £36.93 £480.09 2mg £3.58 £1.13 £2.45 £127.40 7 7 4mg £3.88 £0.86 £3.02 £157.04 Amias® tablets 8mg £9.89 Candesartan tablets £1.53 £8.36 £108.68 16mg 28 £12.72 28 £1.57 £11.15 £144.95 32mg £16.13 £1.97 £14.16 £184.08 75mg £9.69 £3.07 £6.62 £86.06 Aprovel® tablets 150mg 28 £11.84 Irbesartan tablets 28 £5.78 £6.06 £78.78 300mg £15.93 £5.84 £10.09 £131.17 10mg £51.13 Leflunomide tablets 10mg £4.79 £46.34 £556.08 Arava® tablets 30 30 20mg £61.36 Leflunomide tablets 20mg £4.83 £56.53 £678.36 30mg £13.99 £4.68 £9.31 £121.03 60mg £20.11 £3.03 £17.08 £222.04 Arcoxia® tablets 28 Etoricoxib tablets 28 90mg £22.96 £2.99 £19.97 £259.61 120mg £24.11 £12.35 £11.76 £1,328.88 5mg £59.85 £1.38 £58.47 £760.11 Aricept® tablets 28 Donepezil tablets 28 10mg £83.89 £2.42 £57.43 £746.59 5mg £59.85 Donepezil orodispersible sugar free £9.38 £50.47 £656.11 Aricept Evess® tablets 28 28 10mg £83.89 tablets £10.43 £73.46 £954.98 Arimidex® tablets 1mg 28 £68.56 Anastrazole tablets 28 £1.85 £66.71 £867.23 Aromasin® tablets 25mg 30 £88.80 Exemestane tablets 30 £33.52 £55.28 £663.36 5mg £96.04 £1.38 £94.66 £1,230.58 10mg £96.04 £1.57 £94.47 £1,228.11 Arpoya® tablets 28 Aripiprazole tablets 28 15mg £96.04 £1.74 £94.30 £1,225.90 30mg £192.08 £13.57 £178.51 £2,320.63 Audmonal® capsules £14.80 -

CCA Senior Care Options Formulary
Commonwealth Care Alliance Senior Care Option HMO SNP 2021 List of Covered Drugs Formulary 30 Winter Street • Boston, MA 02108 PLEASE READ: THIS DOCUMENT CONTAINS INFORMATION ABOUT THE DRUGS WE COVER IN THIS PLAN This formulary was updated on 06/01/2021. For more recent information or other questions, please contact Senior Care Options Program (HMO SNP) Member Services, at 1-866-610-2273 or, for TTY users, 711, 8 a.m. – 8 p.m., 7 days a week, or visit www.commonwealthcaresco.org. HPMS Approved Formulary File Submission ID 00021589, Version Number 11 Senior Care Options Program (HMO SNP) 2021 Formulary (List of Covered Drugs) PLEASE READ: THIS DOCUMENT CONTAINS INFORMATION ABOUT THE DRUGS WE COVER IN THIS PLAN HPMS Approved Formulary File Submission ID 00021589, Version Number 11 Note to existing members: This formulary has changed since last year. Please review this document to make sure that it still contains the drugs you take. When this drug list (formulary) refers to “we,” “us”, or “our,” it means Commonwealth Care Alliance. When it refers to “plan” or “our plan,” it means 2021 Senior Care Options Program. This document includes list of the drugs (formulary) for our plan which is current as of 06/01/2021. This formulary document applies to all SCO members. For an updated formulary, please contact us. Our contact information, along with the date we last updated the formulary, appears on the front and back cover pages. You must generally use network pharmacies to use your prescription drug benefit. Benefits, formulary, pharmacy network, and/or copayments/coinsurance may change on January 1, 2022, and from time to time during the year. -

Information for the Patient <Ibandronic Acid 50 Mg Film-Coated Tablets>
NL/H/1834/001-002/DCIbandronic acid 50 mg PL, NL1834 Ibandronic acid 50 mg / 150 mg film-coated tablets Submission: 12/2016 Package leaflet: Information for the patient <Ibandronic acid 50 mg film-coated tablets> Ibandronic acid Read all of this leaflet carefully before you start taking this medicine because it contains important information for you. - Keep this leaflet. You may need to read it again. - If you have any further questions, ask your doctor or pharmacist. - This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their signs of illness are the same as yours. - If you get any side effects, talk to your doctor or pharmacist. This includes any possible side effects not listed in this leaflet. See section 4. What is in this leaflet 1. What <Ibandronic acid 50 mg> is and what it is used for. 2. What you need to know before you take <Ibandronic acid 50 mg>. 3. How to take <Ibandronic acid 50 mg>. 4. Possible side effects. 5. How to store <Ibandronic acid 50 mg> 6. Contents of the pack and other information. 1. What <Ibandronic acid 50 mg> is and what it is used for <Ibandronic acid 50 mg> contains the active substance ibandronic acid. This belongs to a group of medicines called bisphosphonates. <Ibandronic acid 50 mg> is used in adults and prescribed to you if you have breast cancer that has spread to your bones (called bone “metastases”). It helps to prevent your bones from breaking (fractures). -

Ibandronic Acid for Breast Cancer
Ibandronic Acid for Breast Cancer Ibandronic acid is a bisphosphonate. This treatment works by slowing down the rate of bone change. There are two types of bone cells: osteoclasts which break down old bone and osteoblasts which build up new bone. Ibandronic acid stops osteoclast bone resorption and reduces bone mineral loss. This treatment helps to ensure the balance of these cells is correct and bone stays healthy. Bisphosphonates are used in the treatment of osteoporosis. They have also been used for many years in patients with advanced or secondary breast cancer to treat breast cancer that has spread to the bones. Because of this, several clinical trials were done in patients with early breast cancer to see if bisphosphonates can prevent the development of secondary breast cancer to the bones and other sites. Although these trials did not individually show any clear benefit, in 2015 a study was published in which the individual patient data from all the trials were combined showed a statistically and clinically meaningful improvement in breast cancer survival for post-menopausal women. Your treatment Your doctor or non-medical prescriber has prescribed Ibandronic acid for you. The treatment will be an oral tablet (taken by mouth) at a dose of 50mg once a day. It is taken every day for the maximum duration of three years. This treatment can have serious side-effects. It is very important that you report side effects straight away. Don’t delay, if you feel unwell, please ring Triage line 01793 604348 (in hours Monday to Friday 08.30-17.30) 01793 604400 (out of hours) How to take your medication Take your tablet in the morning, after an overnight fast (at least 6 hours) and before the first food or drink of the day.