Subcellular Ca2+ Signaling in the Heart: the Role of Ryanodine Receptor Sensitivity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Calcium-Induced Calcium Release in Smooth Muscle7 Loose Coupling
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central Calcium-induced Calcium Release in Smooth Muscle✪ Loose Coupling between the Action Potential and Calcium Release M.L. Collier, G. Ji, Y.-X. Wang, and M.I. Kotlikoff From the Department of Animal Biology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6046 abstract Calcium-induced calcium release (CICR) has been observed in cardiac myocytes as elementary cal- cium release events (calcium sparks) associated with the opening of L-type Ca2ϩ channels. In heart cells, a tight coupling between the gating of single L-type Ca2ϩ channels and ryanodine receptors (RYRs) underlies calcium re- lease. Here we demonstrate that L-type Ca2ϩ channels activate RYRs to produce CICR in smooth muscle cells in the form of Ca2ϩ sparks and propagated Ca2ϩ waves. However, unlike CICR in cardiac muscle, RYR channel open- ing is not tightly linked to the gating of L-type Ca2ϩ channels. L-type Ca2ϩ channels can open without triggering Ca2ϩ sparks and triggered Ca2ϩ sparks are often observed after channel closure. CICR is a function of the net flux of Ca2ϩ ions into the cytosol, rather than the single channel amplitude of L-type Ca2ϩ channels. Moreover, unlike CICR in striated muscle, calcium release is completely eliminated by cytosolic calcium buffering. Thus, L-type Ca2ϩ channels are loosely coupled to RYR through an increase in global [Ca2ϩ] due to an increase in the effective distance between L-type Ca2ϩ channels and RYR, resulting in an uncoupling of the obligate relationship that exists in striated muscle between the action potential and calcium release. -

Microarchitecture of the Dyad
Cardiovascular Research (2013) 98, 169–176 SPOTLIGHT REVIEW doi:10.1093/cvr/cvt025 Microarchitecture of the dyad David R.L. Scriven, Parisa Asghari, and Edwin D.W. Moore* Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3 Received 12 December 2012; revised 2 February 2013; accepted 4 February 2013; online publish-ahead-of-print 11 February 2013 Downloaded from https://academic.oup.com/cardiovascres/article/98/2/169/278625 by guest on 23 September 2021 Abstract This review highlights recent and ongoing discoveries that are transforming the previously held view of dyad structure and function. New data show that dyads vary greatly in both structure and in their associated molecules. Dyads can contain varying numbers of type 2 ryanodine receptor (RYR2) clusters that range in size from one to hundreds of tetramers and they can adopt numerous orientations other than the expected checkerboard. The association of Cav1.2 with RYR2, which defines the couplon, is not absolute, leading to a number of scenarios such as dyads without couplons and those in which only a fraction of the clusters are in couplons. Different dyads also vary in the transporters and exchangers with which they are associated producing functional differences that amplify their structural diversity. The essential role of proteins, such as junctophilin-2, calsequestrin, triadin, and junctin that main- tain both the functional and structural integrity of the dyad have recently been elucidated giving a new mechanistic understanding of heart diseases, such as arrhythmias, hypertension, failure, and sudden cardiac death. -

Calcium Signaling and Cardiac Arrhythmias
Review Calcium Signaling Series Donald M. Bers, Guest Editor Calcium Signaling and Cardiac Arrhythmias Andrew P. Landstrom, Dobromir Dobrev, Xander H.T. Wehrens Abstract: There has been a significant progress in our understanding of the molecular mechanisms by which calcium (Ca2+) ions mediate various types of cardiac arrhythmias. A growing list of inherited gene defects can cause potentially lethal cardiac arrhythmia syndromes, including catecholaminergic polymorphic ventricular Downloaded from tachycardia, congenital long QT syndrome, and hypertrophic cardiomyopathy. In addition, acquired deficits of multiple Ca2+-handling proteins can contribute to the pathogenesis of arrhythmias in patients with various types of heart disease. In this review article, we will first review the key role of Ca2+ in normal cardiac function—in particular, excitation–contraction coupling and normal electric rhythms. The functional involvement of Ca2+ in distinct arrhythmia mechanisms will be discussed, followed by various inherited arrhythmia syndromes caused 2+ http://circres.ahajournals.org/ by mutations in Ca -handling proteins. Finally, we will discuss how changes in the expression of regulation of Ca2+ channels and transporters can cause acquired arrhythmias, and how these mechanisms might be targeted for therapeutic purposes. (Circ Res. 2017;120:1969-1993. DOI: 10.1161/CIRCRESAHA.117.310083.) Key Words: arrhythmias, cardiac ■ atrial fibrillation ■ calcium channels ■ cardiomyopathy ■ ryanodine receptor calcium release channel 2+ by guest on June 11, 2017 he bivalent cation calcium (Ca ) represents one of the Overview of Excitation–Contraction Tmost ubiquitous signal transduction molecules known.1 Coupling in the Heart It mediates a diverse array of biological functions including Regular contraction of the heart requires the conversion of muscle contraction, cellular exocytosis, neuronal activity, and electric activation (excitation) into mechanical force (con- triggering of programmed cell death. -
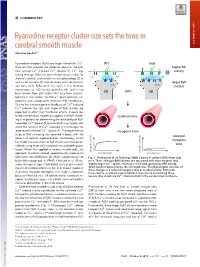
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig. -

An Investigative Mathematical Model of Calcium-Induced Calcium Release
Biophysical Journal Volume 83 July 2002 59–78 59 Termination of Cardiac Ca2؉ Sparks: An Investigative Mathematical Model of Calcium-Induced Calcium Release Eric A. Sobie,* Keith W. Dilly,* Jader dos Santos Cruz,* W. Jonathan Lederer,* and M. Saleet Jafri† *Medical Biotechnology Center, University of Maryland Biotechnology Center, Baltimore, Maryland 21201, and †Department of Mathematical Sciences, The University of Texas at Dallas, Richardson, Texas 75083 USA ABSTRACT A Ca2ϩ spark arises when a cluster of sarcoplasmic reticulum (SR) channels (ryanodine receptors or RyRs) opens to release calcium in a locally regenerative manner. Normally triggered by Ca2ϩ influx across the sarcolemmal or transverse tubule membrane neighboring the cluster, the Ca2ϩ spark has been shown to be the elementary Ca2ϩ signaling event of excitation–contraction coupling in heart muscle. However, the question of how the Ca2ϩ spark terminates remains a central, unresolved issue. Here we present a new model, “sticky cluster,” of SR Ca2ϩ release that simulates Ca2ϩ spark behavior and enables robust Ca2ϩ spark termination. Two newly documented features of RyR behavior have been incorpo- rated in this otherwise simple model: “coupled gating” and an opening rate that depends on SR lumenal [Ca2ϩ]. Using a Monte Carlo method, local Ca2ϩ-induced Ca2ϩ release from clusters containing between 10 and 100 RyRs is modeled. After release is triggered, Ca2ϩ flux from RyRs diffuses into the cytosol and binds to intracellular buffers and the fluorescent Ca2ϩ indicator fluo-3 to produce the model Ca2ϩ spark. Ca2ϩ sparks generated by the sticky cluster model resemble those observed experimentally, and Ca2ϩ spark duration and amplitude are largely insensitive to the number of RyRs in a cluster. -

Defects in T-Tubular Electrical Activity Underlie Local Alterations of Calcium Release in Heart Failure
Defects in T-tubular electrical activity underlie local alterations of calcium release in heart failure Claudia Crocinia, Raffaele Coppinib, Cecilia Ferrantinic, Ping Yand, Leslie M. Loewd, Chiara Tesic, Elisabetta Cerbaib, Corrado Poggesic, Francesco S. Pavonea,e,f, and Leonardo Sacconia,f,1 aEuropean Laboratory for Non-Linear Spectroscopy, 50019 Florence, Italy; bDivision of Pharmacology, Department “NeuroFarBa,” University of Florence, 50139 Florence, Italy; cDivision of Physiology, Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy; dR. D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center, Farmington, CT 06030; eDepartment of Physics and Astronomy, University of Florence, 50019 Sesto Fiorentino, Italy; and fNational Institute of Optics, National Research Council, 50125 Florence, Italy Edited by Clara Franzini-Armstrong, University of Pennsylvania Medical Center, Philadelphia, PA, and approved September 15, 2014 (received for review June 20, 2014) Action potentials (APs), via the transverse axial tubular system in a rat model of postischemic HF, structurally remodeled TATS + (TATS), synchronously trigger uniform Ca2 release throughout the exhibits abnormal electrical activity, i.e., failure of AP propagation cardiomyocyte. In heart failure (HF), TATS structural remodeling and presence of local spontaneous depolarizations. Tubular AP occurs, leading to asynchronous Ca2+ release across the myocyte failures and spontaneous activity can potentially aggravate asyn- + and contributing to contractile dysfunction. In cardiomyocytes from chronous Ca2 release and determine nonhomogeneous myofibril + failing rat hearts, we previously documented the presence of TATS contraction. Simultaneous recording of local Ca2 release and AP in elements which failed to propagate AP and displayed spontaneous the tubular network is needed to unravel the consequences of these + + electrical activity; the consequence for Ca2 release remained, how- electrical anomalies on intracellular Ca2 dynamics. -

Paradoxical Buffering of Calcium by Calsequestrin Demonstrated for the Calcium Store of Skeletal Muscle
A r t i c l e Paradoxical buffering of calcium by calsequestrin demonstrated for the calcium store of skeletal muscle Leandro Royer,1 Monika Sztretye,1 Carlo Manno,1 Sandrine Pouvreau,1 Jingsong Zhou,1 Bjorn C. Knollmann,2 Feliciano Protasi,3 Paul D. Allen,4 and Eduardo Ríos1 1Section of Cellular Signaling, Department of Molecular Biophysics and Physiology, Rush University, Chicago, IL 60612 2Department of Medicine and Pharmacology, Vanderbilt University, Nashville, TN 37240 3Centro Scienze dell’Invecchiamento, Università G. d’Annunzio, 66100 Chieti, Italy 4Department of Anesthesia, Perioperative and Pain Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115 Contractile activation in striated muscles requires a Ca2+ reservoir of large capacity inside the sarcoplasmic reticu- lum (SR), presumably the protein calsequestrin. The buffering power of calsequestrin in vitro has a paradoxical dependence on [Ca2+] that should be valuable for function. Here, we demonstrate that this dependence is present in living cells. Ca2+ signals elicited by membrane depolarization under voltage clamp were compared in single skel- etal fibers of wild-type (WT) and double (d) Casq-null mice, which lack both calsequestrin isoforms. In nulls, Ca2+ release started normally, but the store depleted much more rapidly than in the WT. This deficit was reflected in the evolution of SR evacuability, E, which is directly proportional to SR Ca2+ permeability and inversely to its Ca2+ buff- ering power, B. In WT mice E starts low and increases progressively as the SR is depleted. In dCasq-nulls, E started high and decreased upon Ca2+ depletion. An elevated E in nulls is consistent with the decrease in B expected upon deletion of calsequestrin. -

The L-Type Calcium Channel in the Heart: the Beat Goes On
The L-type calcium channel in the heart: the beat goes on Ilona Bodi, … , Shahab A. Akhter, Arnold Schwartz J Clin Invest. 2005;115(12):3306-3317. https://doi.org/10.1172/JCI27167. Review Sydney Ringer would be overwhelmed today by the implications of his simple experiment performed over 120 years ago showing that the heart would not beat in the absence of Ca2+. Fascination with the role of Ca2+ has proliferated into all aspects of our understanding of normal cardiac function and the progression of heart disease, including induction of cardiac hypertrophy, heart failure, and sudden death. This review examines the role of Ca2+ and the L-type voltage- dependent Ca2+ channels in cardiac disease. Find the latest version: https://jci.me/27167/pdf Review The L-type calcium channel in the heart: the beat goes on Ilona Bodi, Gabor Mikala, Sheryl E. Koch, Shahab A. Akhter, and Arnold Schwartz Institute of Molecular Pharmacology and Biophysics, University of Cincinnati College of Medicine, Department of Surgery, Cincinnati, Ohio, USA. Sydney Ringer would be overwhelmed today by the implications of his simple experiment performed over 120 years ago showing that the heart would not beat in the absence of Ca2+. Fascination with the role of Ca2+ has pro- liferated into all aspects of our understanding of normal cardiac function and the progression of heart disease, including induction of cardiac hypertrophy, heart failure, and sudden death. This review examines the role of Ca2+ and the L-type voltage-dependent Ca2+ channels in cardiac disease. When Sydney Ringer (1) discovered the vital role of Ca2+ in the exchanger (NCX). -

Markov Chain Models of Calcium Puffs and Sparks
W&M ScholarWorks Dissertations, Theses, and Masters Projects Theses, Dissertations, & Master Projects 2008 Markov chain models of calcium puffs and sparks Jeffrey R. Groff College of William & Mary - Arts & Sciences Follow this and additional works at: https://scholarworks.wm.edu/etd Part of the Biophysics Commons Recommended Citation Groff, Jeffrey R., "Markov chain models of calcium puffs and sparks" (2008). Dissertations, Theses, and Masters Projects. Paper 1539623333. https://dx.doi.org/doi:10.21220/s2-yn0e-1d19 This Dissertation is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Dissertations, Theses, and Masters Projects by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. Markov Chain Models of Calcium Puffs and Sparks Jeffrey R. Groff Newport News, Virginia Master of Science, The College of William & Mary, 2004 Bachelor of Arts, McDaniel College, 2001 A Dissertation presented to the Graduate Faculty of the College of William and Mary in Candidacy for the Degree of Doctor of Philosophy Department of Applied Science The College of William and Mary January, 2008 APPROVAL PAGE This Dissertation is submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Approved by the Comm:z~er, 2007 Committee Chair Dr. Gr gory D. Smith, Applied Science The College of William & Mary Dr. Christopher A. Del Negro, plied Science The College of William & Mary Dr. Mark K. Hinders, AppliedScience The College of William & Mary Dr. Eric A. Sobie, Pharmacology and Systems Therapeutics Mount Sinai School of Medicine ABSTRACT PAGE Localized cytosolic Ca2+ elevations known as puffs and sparks are important regulators of cellular function that arise due to the cooperative activity of Ca 2+-regulated inositol 1 ,4,5-trisphosphate receptors (IP3Rs) or ryanodine receptors (RyRs) co-localized at Ca2+ release sites on the surface of the endoplasmic reticulum or sarcoplasmic reticulum. -

Calcium Homeostasis Is Modified in Skeletal Muscle Fibers of Small
International Journal of Molecular Sciences Article Calcium Homeostasis Is Modified in Skeletal Muscle Fibers of Small Ankyrin1 Knockout Mice 1, 2, 2,3 2 2 Enrico Pierantozzi y ,Péter Szentesi y ,Dána Al-Gaadi , Tamás Oláh , Beatrix Dienes , Mónika Sztretye 2, Daniela Rossi 1, Vincenzo Sorrentino 1 and László Csernoch 2,* 1 Department of Molecular and Developmental Medicine, Molecular Medicine Section, University of Siena, 53100 Siena, Italy 2 Department of Physiology, Medical Faculty, University of Debrecen, H-4002 Debrecen, Hungary 3 Doctoral School of Molecular Medicine, University of Debrecen, H-4002 Debrecen, Hungary * Correspondence: [email protected] These authors contribute equally to this work. y Received: 8 May 2019; Accepted: 4 July 2019; Published: 9 July 2019 Abstract: Small Ankyrins (sAnk1) are muscle-specific isoforms generated by the Ank1 gene that participate in the organization of the sarcoplasmic reticulum (SR) of striated muscles. Accordingly, the volume of SR tubules localized around the myofibrils is strongly reduced in skeletal muscle fibers of 4- and 10-month-old sAnk1 knockout (KO) mice, while additional structural alterations only develop with aging. To verify whether the lack of sAnk1 also alters intracellular Ca2+ handling, cytosolic Ca2+ levels were analyzed in stimulated skeletal muscle fibers from 4- and 10-month-old sAnk1 KO mice. The SR Ca2+ content was reduced in sAnk1 KO mice regardless of age. The amplitude of the Ca2+ transients induced by depolarizing pulses was decreased in myofibers of sAnk1 KO with respect to wild type (WT) fibers, while their voltage dependence was not affected. Furthermore, analysis of spontaneous Ca2+ release events (sparks) on saponin-permeabilized muscle fibers indicated that the frequency of sparks was significantly lower in fibers from 4-month-old KO mice compared to WT. -

Coming of Age Or Midlife Crisis? Erick O
Hernández-Ochoa and Schneider Skeletal Muscle (2018) 8:22 https://doi.org/10.1186/s13395-018-0167-9 REVIEW Open Access Voltage sensing mechanism in skeletal muscle excitation-contraction coupling: coming of age or midlife crisis? Erick O. Hernández-Ochoa and Martin F. Schneider* Abstract The process by which muscle fiber electrical depolarization is linked to activation of muscle contraction is known as excitation-contraction coupling (ECC). Our understanding of ECC has increased enormously since the early scientific descriptions of the phenomenon of electrical activation of muscle contraction by Galvani that date back to the end of the eighteenth century. Major advances in electrical and optical measurements, including muscle fiber voltage clamp to reveal membrane electrical properties, in conjunction with the development of electron microscopy to unveil structural details provided an elegant view of ECC in skeletal muscle during the last century. This surge of knowledge on structural and biophysical aspects of the skeletal muscle was followed by breakthroughs in biochemistry and molecular biology, which allowed for the isolation, purification, and DNA sequencing of the muscle fiber membrane calcium channel/transverse tubule (TT) membrane voltage sensor (Cav1.1) for ECC and of the muscle ryanodine receptor/sarcoplasmic reticulum Ca2+ release channel (RyR1), two essential players of ECC in skeletal muscle. In regard to the process of voltage sensing for controlling calcium release, numerous studies support the concept that the TT Cav1.1 channel is the voltage sensor for ECC, as well as also being aCa2+ channel in the TT membrane. In this review, we present early and recent findings that support and define the role of Cav1.1 as a voltage sensor for ECC.