The Control of Formation and Recovery of Transverse Tubules in the Heart
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Calcium-Induced Calcium Release in Smooth Muscle7 Loose Coupling
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central Calcium-induced Calcium Release in Smooth Muscle✪ Loose Coupling between the Action Potential and Calcium Release M.L. Collier, G. Ji, Y.-X. Wang, and M.I. Kotlikoff From the Department of Animal Biology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6046 abstract Calcium-induced calcium release (CICR) has been observed in cardiac myocytes as elementary cal- cium release events (calcium sparks) associated with the opening of L-type Ca2ϩ channels. In heart cells, a tight coupling between the gating of single L-type Ca2ϩ channels and ryanodine receptors (RYRs) underlies calcium re- lease. Here we demonstrate that L-type Ca2ϩ channels activate RYRs to produce CICR in smooth muscle cells in the form of Ca2ϩ sparks and propagated Ca2ϩ waves. However, unlike CICR in cardiac muscle, RYR channel open- ing is not tightly linked to the gating of L-type Ca2ϩ channels. L-type Ca2ϩ channels can open without triggering Ca2ϩ sparks and triggered Ca2ϩ sparks are often observed after channel closure. CICR is a function of the net flux of Ca2ϩ ions into the cytosol, rather than the single channel amplitude of L-type Ca2ϩ channels. Moreover, unlike CICR in striated muscle, calcium release is completely eliminated by cytosolic calcium buffering. Thus, L-type Ca2ϩ channels are loosely coupled to RYR through an increase in global [Ca2ϩ] due to an increase in the effective distance between L-type Ca2ϩ channels and RYR, resulting in an uncoupling of the obligate relationship that exists in striated muscle between the action potential and calcium release. -

Cardiac Physiologyc As a Country
Type to enter text CardiacCardiovascular PhysiologyC as a Physiology Country Doc EpisodeEpisode 2:2: The EKG Electrocardiography I Patrick Eggena, M.D. Novateur Medmedia Type EPISODEto enter text 2 Type to enter text THE EKG by Patrick Eggena, M.D. i Copyright This Episode is derived from: Course in Cardiovascular Physiology by Patrick Eggena, M.D. © Copyright Novateur Medmedia, LLC. April 13, 2012 The United States Copyright Registration Number: PAu3-662-048 Ordering Information via iBooks: ISBN 978-0-9905771-1-9 Cardiac Physiology as a Country Doc, Episode 2: The EKG. Contact Information: Novateur Medmedia, LLC 39 Terry Hill Road, Carmel, NY 10512 email: [email protected] Credits: Oil Paintings by Bonnie Eggena, PsD. Music by Alan Goodman from his CD Under the Bed, Cancoll Music, copyright 2005 (with permission). Illustrations, movies, text, and lectures by Patrick Eggena, M.D. Note: Knowledge in the basic and clinical sciences is constantly changing. The reader is advised to carefully consult the instruc- tions and informational material included in the package inserts of each drug or therapeutic agent before administration. The Country Doctor Series illustrates Physiological Principles and is not intended as a guide to Medical Therapeutics. Care has been taken to present correct information in this book, however, the author and publisher are not responsible for errors or omissions or for any consequence from application of the information in this work and make no warranty, expressed or implied, with re- spect to the contents of this publication or that its operation will be uninterrupted and error free on any particular recording de- vice. -

Microarchitecture of the Dyad
Cardiovascular Research (2013) 98, 169–176 SPOTLIGHT REVIEW doi:10.1093/cvr/cvt025 Microarchitecture of the dyad David R.L. Scriven, Parisa Asghari, and Edwin D.W. Moore* Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3 Received 12 December 2012; revised 2 February 2013; accepted 4 February 2013; online publish-ahead-of-print 11 February 2013 Downloaded from https://academic.oup.com/cardiovascres/article/98/2/169/278625 by guest on 23 September 2021 Abstract This review highlights recent and ongoing discoveries that are transforming the previously held view of dyad structure and function. New data show that dyads vary greatly in both structure and in their associated molecules. Dyads can contain varying numbers of type 2 ryanodine receptor (RYR2) clusters that range in size from one to hundreds of tetramers and they can adopt numerous orientations other than the expected checkerboard. The association of Cav1.2 with RYR2, which defines the couplon, is not absolute, leading to a number of scenarios such as dyads without couplons and those in which only a fraction of the clusters are in couplons. Different dyads also vary in the transporters and exchangers with which they are associated producing functional differences that amplify their structural diversity. The essential role of proteins, such as junctophilin-2, calsequestrin, triadin, and junctin that main- tain both the functional and structural integrity of the dyad have recently been elucidated giving a new mechanistic understanding of heart diseases, such as arrhythmias, hypertension, failure, and sudden cardiac death. -

Insights from Computational Modeling Into the Contribution of Mechano-Calcium Feedback on the Cardiac End-Systolic Force-Length Relationship
fphys-11-00587 May 28, 2020 Time: 19:46 # 1 ORIGINAL RESEARCH published: 29 May 2020 doi: 10.3389/fphys.2020.00587 Insights From Computational Modeling Into the Contribution of Mechano-Calcium Feedback on the Cardiac End-Systolic Force-Length Relationship Megan E. Guidry1, David P. Nickerson1, Edmund J. Crampin2,3, Martyn P. Nash1,4, Denis S. Loiselle1,5 and Kenneth Tran1* 1 Auckland Bioengineering Institute, The University of Auckland, Auckland, New Zealand, 2 Systems Biology Laboratory, School of Mathematics and Statistics, The University of Melbourne, Melbourne, VIC, Australia, 3 ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, School of Chemical and Biomedical Engineering, The University of Melbourne, Melbourne, VIC, Australia, 4 Department of Engineering Science, The University of Auckland, Auckland, Edited by: New Zealand, 5 Department of Physiology, The University of Auckland, Auckland, New Zealand Leonid Katsnelson, Institute of Immunology and Physiology (RAS), Russia In experimental studies on cardiac tissue, the end-systolic force-length relation (ESFLR) Reviewed by: has been shown to depend on the mode of contraction: isometric or isotonic. The Andrey K. Tsaturyan, Lomonosov Moscow State University, isometric ESFLR is derived from isometric contractions spanning a range of muscle Russia lengths while the isotonic ESFLR is derived from shortening contractions across a Aurore Lyon, range of afterloads. The ESFLR of isotonic contractions consistently lies below its Maastricht University, Netherlands isometric counterpart. Despite the passing of over a hundred years since the first insight *Correspondence: Kenneth Tran by Otto Frank, the mechanism(s) underlying this protocol-dependent difference in the [email protected] ESFLR remain incompletely explained. -

Strategic Review of Cardiac Physiology Services in England: Final Report
STRATEGIC REVIEW OF CARDIAC PHYSIOLOGY SERVICES IN ENGLAND: FINAL REPORT Strategic Review of Cardiac Physiology Services in England Page 1 Final Report: 12/05/2015 CONTENTS Page Foreword 3 Summary of recommendations 4 Background 7 Purpose of the Review 8 Working arrangements and governance 8 Delivering a service model for the future 8 Delivering a cardiac physiology service to quality standards 16 Developing a cardiac physiology workforce for the future 18 Commissioning for quality 20 Appendix 1: 23 Cardiac physiology services in summary Appendix 2: 27 Working arrangements and governance Appendix 3: 28 Award in Practical Electrocardiography (APECG) Appendix 4: 29 Proposed possible key performance indicators Appendix 5: 30 Current status of standards development across a range of cardiac physiology investigations Appendix 6: 32 Current training, accreditation and standards Appendix 7: 33 National Review of Cardiac Physiology Services - Modelling the Future Workforce References 34 Glossary 36 Acknowledgements 38 Strategic Review of Cardiac Physiology Services in England Page 2 Final Report: 12/05/2015 FOREWORD In 2013, the Department of Health published the Cardiovascular Outcomes Strategy, a call to action to the health and care systems, which set out ten key actions to deliver for patients with cardiovascular disease improvements in their health outcomes. Cardiac physiology services are key to delivering the improved treatment and care, which is the ambition of the Outcomes Strategy. This Strategic Review of Cardiac Physiology Services was set up to make recommendations to NHS England, Health Education England and across the health and care system about the innovations in service delivery, models of care and workforce configuration which are needed to transform patients’ experience of cardiovascular disease care. -

Cardiac Physiology
Chapter 20 Cardiac Physiology LENA S. SUN • JOHANNA SCHWARZENBERGER • RADHIKA DINAVAHI K EY P OINTS • The cardiac cycle is the sequence of electrical and mechanical events during the course of a single heartbeat. • Cardiac output is determined by the heart rate, myocardial contractility, and preload and afterload. • The majority of cardiomyocytes consist of myofibrils, which are rodlike bundles that form the contractile elements within the cardiomyocyte. • The basic working unit of contraction is the sarcomere. • Gap junctions are responsible for the electrical coupling of small molecules between cells. • Action potentials have four phases in the heart. • The key player in cardiac excitation-contraction coupling is the ubiquitous second messenger calcium. • Calcium-induced sparks are spatially and temporally patterned activations of localized calcium release that are important for excitation-contraction coupling and regulation of automaticity and contractility. • β -Adrenoreceptors stimulate chronotropy, inotropy, lusitropy, and dromotropy. • Hormones with cardiac action can be synthesized and secreted by cardiomyocytes or produced by other tissues and delivered to the heart. • Cardiac reflexes are fast-acting reflex loops between the heart and central nervous system that contribute to the regulation of cardiac function and the maintenance of physiologic homeostasis. In 1628, English physician, William Harvey, first advanced oxygen (O2) and nutrients and to remove carbon dioxide the modern concept of circulation with the heart as the (CO2) and metabolites from various tissue beds. generator for the circulation. Modern cardiac physiology includes not only physiology of the heart as a pump but also concepts of cellular and molecular biology of the car- PHYSIOLOGY OF THE INTACT HEART diomyocyte and regulation of cardiac function by neural and humoral factors. -

Webinar Ch 8
Animal Anatomy and Physiology 1 Webinar Chapter 8 Cardiovascular System The Cardiovascular System Chapter 8 Pages 205-219 Textbook Learning Objectives Chapter 8 – Page 205 • List and describe the layers of the heart wall • List the chambers of the heart and describe the path of blood flow through the heart • Describe the structure and locations of the heart valves • Differentiate between systole and diastole • Describe the process of depolarization and repolarization of cardiac muscle cells • Describe the pathway of the electrical impulse generated by the SA node • List and describe the unique anatomical features of the fetal circulatory system • List the events responsible for the heart sounds heard on auscultation • List the factors that influence heart rate and cardiac output • Describe the relationship between cardiac output, heart rate, and stroke volume • Describe the structures of arteries, capillaries, and veins • List the major arteries and veins that travel from the heart to the systemic circulation • List the names and locations of veins commonly used for venipuncture in animals Cardiovascular System Heart Blood Vessels Blood YourDogsHeart.com http://www.yourdogsheart.com/ Your Cat’s Heart http://www.pethealthnetwork.com/cat-health/your- cat%E2%80%99s-heart-feline-heart-disease Great Human Heart A&P website http://www.nhlbi.nih.gov/health/health-topics/topics/hhw/ Internal Medicine The KEY Is Cellular Health Cellular Health • Healthy Cells = Health Animal Body • Diseased Cells = Diseased Animal Body • Too Many Diseased Cells -

Calcium Signaling and Cardiac Arrhythmias
Review Calcium Signaling Series Donald M. Bers, Guest Editor Calcium Signaling and Cardiac Arrhythmias Andrew P. Landstrom, Dobromir Dobrev, Xander H.T. Wehrens Abstract: There has been a significant progress in our understanding of the molecular mechanisms by which calcium (Ca2+) ions mediate various types of cardiac arrhythmias. A growing list of inherited gene defects can cause potentially lethal cardiac arrhythmia syndromes, including catecholaminergic polymorphic ventricular Downloaded from tachycardia, congenital long QT syndrome, and hypertrophic cardiomyopathy. In addition, acquired deficits of multiple Ca2+-handling proteins can contribute to the pathogenesis of arrhythmias in patients with various types of heart disease. In this review article, we will first review the key role of Ca2+ in normal cardiac function—in particular, excitation–contraction coupling and normal electric rhythms. The functional involvement of Ca2+ in distinct arrhythmia mechanisms will be discussed, followed by various inherited arrhythmia syndromes caused 2+ http://circres.ahajournals.org/ by mutations in Ca -handling proteins. Finally, we will discuss how changes in the expression of regulation of Ca2+ channels and transporters can cause acquired arrhythmias, and how these mechanisms might be targeted for therapeutic purposes. (Circ Res. 2017;120:1969-1993. DOI: 10.1161/CIRCRESAHA.117.310083.) Key Words: arrhythmias, cardiac ■ atrial fibrillation ■ calcium channels ■ cardiomyopathy ■ ryanodine receptor calcium release channel 2+ by guest on June 11, 2017 he bivalent cation calcium (Ca ) represents one of the Overview of Excitation–Contraction Tmost ubiquitous signal transduction molecules known.1 Coupling in the Heart It mediates a diverse array of biological functions including Regular contraction of the heart requires the conversion of muscle contraction, cellular exocytosis, neuronal activity, and electric activation (excitation) into mechanical force (con- triggering of programmed cell death. -

Advanced P Wave Detection in Ecg Signals During Pathology
www.nature.com/scientificreports OPEN Advanced P Wave Detection in Ecg Signals During Pathology: Evaluation in Diferent Arrhythmia Contexts Lucie Maršánová 1*, Andrea Němcová1, Radovan Smíšek1,2, Martin Vítek1 & Lukáš Smital1 Reliable P wave detection is necessary for accurate and automatic electrocardiogram (ECG) analysis. Currently, methods for P wave detection in physiological conditions are well-described and efcient. However, methods for P wave detection during pathology are not generally found in the literature, or their performance is insufcient. This work introduces a novel method, based on a phasor transform, as well as innovative rules that improve P wave detection during pathology. These rules are based on the extraction of a heartbeats’ morphological features and knowledge of heart manifestation during both physiological and pathological conditions. To properly evaluate the performance of the proposed algorithm in pathological conditions, a standard database with a sufcient number of reference P wave positions is needed. However, such a database did not exist. Thus, ECG experts annotated 12 chosen pathological records from the MIT-BIH Arrhythmia Database. These annotations are publicly available via Physionet. The algorithm performance was also validated using physiological records from the MIT-BIH Arrhythmia and QT databases. The results for physiological signals were Se = 98.42% and PP = 99.98%, which is comparable to other methods. For pathological signals, the proposed method reached Se = 96.40% and PP = 85.84%, which greatly outperforms other methods. This improvement represents a huge step towards fully automated analysis systems being respected by ECG experts. These systems are necessary, particularly in the area of long-term monitoring. -
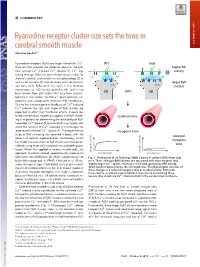
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig. -

An Investigative Mathematical Model of Calcium-Induced Calcium Release
Biophysical Journal Volume 83 July 2002 59–78 59 Termination of Cardiac Ca2؉ Sparks: An Investigative Mathematical Model of Calcium-Induced Calcium Release Eric A. Sobie,* Keith W. Dilly,* Jader dos Santos Cruz,* W. Jonathan Lederer,* and M. Saleet Jafri† *Medical Biotechnology Center, University of Maryland Biotechnology Center, Baltimore, Maryland 21201, and †Department of Mathematical Sciences, The University of Texas at Dallas, Richardson, Texas 75083 USA ABSTRACT A Ca2ϩ spark arises when a cluster of sarcoplasmic reticulum (SR) channels (ryanodine receptors or RyRs) opens to release calcium in a locally regenerative manner. Normally triggered by Ca2ϩ influx across the sarcolemmal or transverse tubule membrane neighboring the cluster, the Ca2ϩ spark has been shown to be the elementary Ca2ϩ signaling event of excitation–contraction coupling in heart muscle. However, the question of how the Ca2ϩ spark terminates remains a central, unresolved issue. Here we present a new model, “sticky cluster,” of SR Ca2ϩ release that simulates Ca2ϩ spark behavior and enables robust Ca2ϩ spark termination. Two newly documented features of RyR behavior have been incorpo- rated in this otherwise simple model: “coupled gating” and an opening rate that depends on SR lumenal [Ca2ϩ]. Using a Monte Carlo method, local Ca2ϩ-induced Ca2ϩ release from clusters containing between 10 and 100 RyRs is modeled. After release is triggered, Ca2ϩ flux from RyRs diffuses into the cytosol and binds to intracellular buffers and the fluorescent Ca2ϩ indicator fluo-3 to produce the model Ca2ϩ spark. Ca2ϩ sparks generated by the sticky cluster model resemble those observed experimentally, and Ca2ϩ spark duration and amplitude are largely insensitive to the number of RyRs in a cluster.