The L-Type Calcium Channel in the Heart: the Beat Goes On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Unnatural Verticilide Enantiomer Inhibits Type 2 Ryanodine Receptor-Mediated Calcium Leak and Is Antiarrhythmic
Unnatural verticilide enantiomer inhibits type 2 ryanodine receptor-mediated calcium leak and is antiarrhythmic Suzanne M. Batistea,1, Daniel J. Blackwellb,1, Kyungsoo Kimb,1, Dmytro O. Kryshtalb, Nieves Gomez-Hurtadob, Robyn T. Rebbeckc, Razvan L. Corneac, Jeffrey N. Johnstona,2, and Bjorn C. Knollmannb,2 aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235; bDepartment of Medicine, Vanderbilt University Medical Center, Nashville, TN 37232; and cDepartment of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by Dale L. Boger, The Scripps Research Institute, La Jolla, CA, and approved January 15, 2019 (received for review September 27, 2018) Ca2+ leak via ryanodine receptor type 2 (RyR2) can cause poten- heart diseases associated with both atrial and ventricular arrhyth- tially fatal arrhythmias in a variety of heart diseases and has also mia (9). Mutations in RyR2 and its binding partners, which increase + been implicated in neurodegenerative and seizure disorders, mak- SR Ca2 leak, cause primary atrial and ventricular arrhythmia ing RyR2 an attractive therapeutic target for drug development. syndromes such as catecholaminergic polymorphic ventricular Here we synthesized and investigated the fungal natural product tachycardia (CPVT), providing strong evidence for the mechanistic and known insect RyR antagonist (−)-verticilide and several conge- contribution of RyR2 to arrhythmia risk in humans (10). Further ners to determine their activity against mammalian RyR2. Although support comes from gene-targeted mouse models of CPVT, where + the cyclooligomeric depsipeptide natural product (−)-verticilide had catecholamine-induced spontaneous Ca2 release from the SR no effect, its nonnatural enantiomer [ent-(+)-verticilide] signifi- via RyR2 generates potentially fatal cardiac arrhythmias (11, 12). -

Calcium-Induced Calcium Release in Smooth Muscle7 Loose Coupling
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by PubMed Central Calcium-induced Calcium Release in Smooth Muscle✪ Loose Coupling between the Action Potential and Calcium Release M.L. Collier, G. Ji, Y.-X. Wang, and M.I. Kotlikoff From the Department of Animal Biology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6046 abstract Calcium-induced calcium release (CICR) has been observed in cardiac myocytes as elementary cal- cium release events (calcium sparks) associated with the opening of L-type Ca2ϩ channels. In heart cells, a tight coupling between the gating of single L-type Ca2ϩ channels and ryanodine receptors (RYRs) underlies calcium re- lease. Here we demonstrate that L-type Ca2ϩ channels activate RYRs to produce CICR in smooth muscle cells in the form of Ca2ϩ sparks and propagated Ca2ϩ waves. However, unlike CICR in cardiac muscle, RYR channel open- ing is not tightly linked to the gating of L-type Ca2ϩ channels. L-type Ca2ϩ channels can open without triggering Ca2ϩ sparks and triggered Ca2ϩ sparks are often observed after channel closure. CICR is a function of the net flux of Ca2ϩ ions into the cytosol, rather than the single channel amplitude of L-type Ca2ϩ channels. Moreover, unlike CICR in striated muscle, calcium release is completely eliminated by cytosolic calcium buffering. Thus, L-type Ca2ϩ channels are loosely coupled to RYR through an increase in global [Ca2ϩ] due to an increase in the effective distance between L-type Ca2ϩ channels and RYR, resulting in an uncoupling of the obligate relationship that exists in striated muscle between the action potential and calcium release. -

Microarchitecture of the Dyad
Cardiovascular Research (2013) 98, 169–176 SPOTLIGHT REVIEW doi:10.1093/cvr/cvt025 Microarchitecture of the dyad David R.L. Scriven, Parisa Asghari, and Edwin D.W. Moore* Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC, Canada V6T 1Z3 Received 12 December 2012; revised 2 February 2013; accepted 4 February 2013; online publish-ahead-of-print 11 February 2013 Downloaded from https://academic.oup.com/cardiovascres/article/98/2/169/278625 by guest on 23 September 2021 Abstract This review highlights recent and ongoing discoveries that are transforming the previously held view of dyad structure and function. New data show that dyads vary greatly in both structure and in their associated molecules. Dyads can contain varying numbers of type 2 ryanodine receptor (RYR2) clusters that range in size from one to hundreds of tetramers and they can adopt numerous orientations other than the expected checkerboard. The association of Cav1.2 with RYR2, which defines the couplon, is not absolute, leading to a number of scenarios such as dyads without couplons and those in which only a fraction of the clusters are in couplons. Different dyads also vary in the transporters and exchangers with which they are associated producing functional differences that amplify their structural diversity. The essential role of proteins, such as junctophilin-2, calsequestrin, triadin, and junctin that main- tain both the functional and structural integrity of the dyad have recently been elucidated giving a new mechanistic understanding of heart diseases, such as arrhythmias, hypertension, failure, and sudden cardiac death. -

Prox1regulates the Subtype-Specific Development of Caudal Ganglionic
The Journal of Neuroscience, September 16, 2015 • 35(37):12869–12889 • 12869 Development/Plasticity/Repair Prox1 Regulates the Subtype-Specific Development of Caudal Ganglionic Eminence-Derived GABAergic Cortical Interneurons X Goichi Miyoshi,1 Allison Young,1 Timothy Petros,1 Theofanis Karayannis,1 Melissa McKenzie Chang,1 Alfonso Lavado,2 Tomohiko Iwano,3 Miho Nakajima,4 Hiroki Taniguchi,5 Z. Josh Huang,5 XNathaniel Heintz,4 Guillermo Oliver,2 Fumio Matsuzaki,3 Robert P. Machold,1 and Gord Fishell1 1Department of Neuroscience and Physiology, NYU Neuroscience Institute, Smilow Research Center, New York University School of Medicine, New York, New York 10016, 2Department of Genetics & Tumor Cell Biology, St. Jude Children’s Research Hospital, Memphis, Tennessee 38105, 3Laboratory for Cell Asymmetry, RIKEN Center for Developmental Biology, Kobe 650-0047, Japan, 4Laboratory of Molecular Biology, Howard Hughes Medical Institute, GENSAT Project, The Rockefeller University, New York, New York 10065, and 5Cold Spring Harbor Laboratory, Cold Spring Harbor, New York 11724 Neurogliaform (RELNϩ) and bipolar (VIPϩ) GABAergic interneurons of the mammalian cerebral cortex provide critical inhibition locally within the superficial layers. While these subtypes are known to originate from the embryonic caudal ganglionic eminence (CGE), the specific genetic programs that direct their positioning, maturation, and integration into the cortical network have not been eluci- dated. Here, we report that in mice expression of the transcription factor Prox1 is selectively maintained in postmitotic CGE-derived cortical interneuron precursors and that loss of Prox1 impairs the integration of these cells into superficial layers. Moreover, Prox1 differentially regulates the postnatal maturation of each specific subtype originating from the CGE (RELN, Calb2/VIP, and VIP). -

Supplementary Table S5. Differentially Expressed Gene Lists of PD-1High CD39+ CD8 Tils According to 4-1BB Expression Compared to PD-1+ CD39- CD8 Tils
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Immunother Cancer Supplementary Table S5. Differentially expressed gene lists of PD-1high CD39+ CD8 TILs according to 4-1BB expression compared to PD-1+ CD39- CD8 TILs Up- or down- regulated genes in Up- or down- regulated genes Up- or down- regulated genes only PD-1high CD39+ CD8 TILs only in 4-1BBneg PD-1high CD39+ in 4-1BBpos PD-1high CD39+ CD8 compared to PD-1+ CD39- CD8 CD8 TILs compared to PD-1+ TILs compared to PD-1+ CD39- TILs CD39- CD8 TILs CD8 TILs IL7R KLRG1 TNFSF4 ENTPD1 DHRS3 LEF1 ITGA5 MKI67 PZP KLF3 RYR2 SIK1B ANK3 LYST PPP1R3B ETV1 ADAM28 H2AC13 CCR7 GFOD1 RASGRP2 ITGAX MAST4 RAD51AP1 MYO1E CLCF1 NEBL S1PR5 VCL MPP7 MS4A6A PHLDB1 GFPT2 TNF RPL3 SPRY4 VCAM1 B4GALT5 TIPARP TNS3 PDCD1 POLQ AKAP5 IL6ST LY9 PLXND1 PLEKHA1 NEU1 DGKH SPRY2 PLEKHG3 IKZF4 MTX3 PARK7 ATP8B4 SYT11 PTGER4 SORL1 RAB11FIP5 BRCA1 MAP4K3 NCR1 CCR4 S1PR1 PDE8A IFIT2 EPHA4 ARHGEF12 PAICS PELI2 LAT2 GPRASP1 TTN RPLP0 IL4I1 AUTS2 RPS3 CDCA3 NHS LONRF2 CDC42EP3 SLCO3A1 RRM2 ADAMTSL4 INPP5F ARHGAP31 ESCO2 ADRB2 CSF1 WDHD1 GOLIM4 CDK5RAP1 CD69 GLUL HJURP SHC4 GNLY TTC9 HELLS DPP4 IL23A PITPNC1 TOX ARHGEF9 EXO1 SLC4A4 CKAP4 CARMIL3 NHSL2 DZIP3 GINS1 FUT8 UBASH3B CDCA5 PDE7B SOGA1 CDC45 NR3C2 TRIB1 KIF14 TRAF5 LIMS1 PPP1R2C TNFRSF9 KLRC2 POLA1 CD80 ATP10D CDCA8 SETD7 IER2 PATL2 CCDC141 CD84 HSPA6 CYB561 MPHOSPH9 CLSPN KLRC1 PTMS SCML4 ZBTB10 CCL3 CA5B PIP5K1B WNT9A CCNH GEM IL18RAP GGH SARDH B3GNT7 C13orf46 SBF2 IKZF3 ZMAT1 TCF7 NECTIN1 H3C7 FOS PAG1 HECA SLC4A10 SLC35G2 PER1 P2RY1 NFKBIA WDR76 PLAUR KDM1A H1-5 TSHZ2 FAM102B HMMR GPR132 CCRL2 PARP8 A2M ST8SIA1 NUF2 IL5RA RBPMS UBE2T USP53 EEF1A1 PLAC8 LGR6 TMEM123 NEK2 SNAP47 PTGIS SH2B3 P2RY8 S100PBP PLEKHA7 CLNK CRIM1 MGAT5 YBX3 TP53INP1 DTL CFH FEZ1 MYB FRMD4B TSPAN5 STIL ITGA2 GOLGA6L10 MYBL2 AHI1 CAND2 GZMB RBPJ PELI1 HSPA1B KCNK5 GOLGA6L9 TICRR TPRG1 UBE2C AURKA Leem G, et al. -

Cldn19 Clic2 Clmp Cln3
NewbornDx™ Advanced Sequencing Evaluation When time to diagnosis matters, the NewbornDx™ Advanced Sequencing Evaluation from Athena Diagnostics delivers rapid, 5- to 7-day results on a targeted 1,722-genes. A2ML1 ALAD ATM CAV1 CLDN19 CTNS DOCK7 ETFB FOXC2 GLUL HOXC13 JAK3 AAAS ALAS2 ATP1A2 CBL CLIC2 CTRC DOCK8 ETFDH FOXE1 GLYCTK HOXD13 JUP AARS2 ALDH18A1 ATP1A3 CBS CLMP CTSA DOK7 ETHE1 FOXE3 GM2A HPD KANK1 AASS ALDH1A2 ATP2B3 CC2D2A CLN3 CTSD DOLK EVC FOXF1 GMPPA HPGD K ANSL1 ABAT ALDH3A2 ATP5A1 CCDC103 CLN5 CTSK DPAGT1 EVC2 FOXG1 GMPPB HPRT1 KAT6B ABCA12 ALDH4A1 ATP5E CCDC114 CLN6 CUBN DPM1 EXOC4 FOXH1 GNA11 HPSE2 KCNA2 ABCA3 ALDH5A1 ATP6AP2 CCDC151 CLN8 CUL4B DPM2 EXOSC3 FOXI1 GNAI3 HRAS KCNB1 ABCA4 ALDH7A1 ATP6V0A2 CCDC22 CLP1 CUL7 DPM3 EXPH5 FOXL2 GNAO1 HSD17B10 KCND2 ABCB11 ALDOA ATP6V1B1 CCDC39 CLPB CXCR4 DPP6 EYA1 FOXP1 GNAS HSD17B4 KCNE1 ABCB4 ALDOB ATP7A CCDC40 CLPP CYB5R3 DPYD EZH2 FOXP2 GNE HSD3B2 KCNE2 ABCB6 ALG1 ATP8A2 CCDC65 CNNM2 CYC1 DPYS F10 FOXP3 GNMT HSD3B7 KCNH2 ABCB7 ALG11 ATP8B1 CCDC78 CNTN1 CYP11B1 DRC1 F11 FOXRED1 GNPAT HSPD1 KCNH5 ABCC2 ALG12 ATPAF2 CCDC8 CNTNAP1 CYP11B2 DSC2 F13A1 FRAS1 GNPTAB HSPG2 KCNJ10 ABCC8 ALG13 ATR CCDC88C CNTNAP2 CYP17A1 DSG1 F13B FREM1 GNPTG HUWE1 KCNJ11 ABCC9 ALG14 ATRX CCND2 COA5 CYP1B1 DSP F2 FREM2 GNS HYDIN KCNJ13 ABCD3 ALG2 AUH CCNO COG1 CYP24A1 DST F5 FRMD7 GORAB HYLS1 KCNJ2 ABCD4 ALG3 B3GALNT2 CCS COG4 CYP26C1 DSTYK F7 FTCD GP1BA IBA57 KCNJ5 ABHD5 ALG6 B3GAT3 CCT5 COG5 CYP27A1 DTNA F8 FTO GP1BB ICK KCNJ8 ACAD8 ALG8 B3GLCT CD151 COG6 CYP27B1 DUOX2 F9 FUCA1 GP6 ICOS KCNK3 ACAD9 ALG9 -

Transcriptomic Uniqueness and Commonality of the Ion Channels and Transporters in the Four Heart Chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A
www.nature.com/scientificreports OPEN Transcriptomic uniqueness and commonality of the ion channels and transporters in the four heart chambers Sanda Iacobas1, Bogdan Amuzescu2 & Dumitru A. Iacobas3,4* Myocardium transcriptomes of left and right atria and ventricles from four adult male C57Bl/6j mice were profled with Agilent microarrays to identify the diferences responsible for the distinct functional roles of the four heart chambers. Female mice were not investigated owing to their transcriptome dependence on the estrous cycle phase. Out of the quantifed 16,886 unigenes, 15.76% on the left side and 16.5% on the right side exhibited diferential expression between the atrium and the ventricle, while 5.8% of genes were diferently expressed between the two atria and only 1.2% between the two ventricles. The study revealed also chamber diferences in gene expression control and coordination. We analyzed ion channels and transporters, and genes within the cardiac muscle contraction, oxidative phosphorylation, glycolysis/gluconeogenesis, calcium and adrenergic signaling pathways. Interestingly, while expression of Ank2 oscillates in phase with all 27 quantifed binding partners in the left ventricle, the percentage of in-phase oscillating partners of Ank2 is 15% and 37% in the left and right atria and 74% in the right ventricle. The analysis indicated high interventricular synchrony of the ion channels expressions and the substantially lower synchrony between the two atria and between the atrium and the ventricle from the same side. Starting with crocodilians, the heart pumps the blood through the pulmonary circulation and the systemic cir- culation by the coordinated rhythmic contractions of its upper lef and right atria (LA, RA) and lower lef and right ventricles (LV, RV). -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

Calcium Signaling and Cardiac Arrhythmias
Review Calcium Signaling Series Donald M. Bers, Guest Editor Calcium Signaling and Cardiac Arrhythmias Andrew P. Landstrom, Dobromir Dobrev, Xander H.T. Wehrens Abstract: There has been a significant progress in our understanding of the molecular mechanisms by which calcium (Ca2+) ions mediate various types of cardiac arrhythmias. A growing list of inherited gene defects can cause potentially lethal cardiac arrhythmia syndromes, including catecholaminergic polymorphic ventricular Downloaded from tachycardia, congenital long QT syndrome, and hypertrophic cardiomyopathy. In addition, acquired deficits of multiple Ca2+-handling proteins can contribute to the pathogenesis of arrhythmias in patients with various types of heart disease. In this review article, we will first review the key role of Ca2+ in normal cardiac function—in particular, excitation–contraction coupling and normal electric rhythms. The functional involvement of Ca2+ in distinct arrhythmia mechanisms will be discussed, followed by various inherited arrhythmia syndromes caused 2+ http://circres.ahajournals.org/ by mutations in Ca -handling proteins. Finally, we will discuss how changes in the expression of regulation of Ca2+ channels and transporters can cause acquired arrhythmias, and how these mechanisms might be targeted for therapeutic purposes. (Circ Res. 2017;120:1969-1993. DOI: 10.1161/CIRCRESAHA.117.310083.) Key Words: arrhythmias, cardiac ■ atrial fibrillation ■ calcium channels ■ cardiomyopathy ■ ryanodine receptor calcium release channel 2+ by guest on June 11, 2017 he bivalent cation calcium (Ca ) represents one of the Overview of Excitation–Contraction Tmost ubiquitous signal transduction molecules known.1 Coupling in the Heart It mediates a diverse array of biological functions including Regular contraction of the heart requires the conversion of muscle contraction, cellular exocytosis, neuronal activity, and electric activation (excitation) into mechanical force (con- triggering of programmed cell death. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -
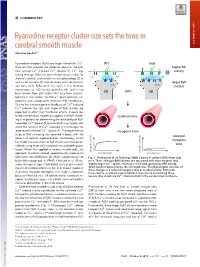
Ryanodine Receptor Cluster Size Sets the Tone in Cerebral Smooth Muscle
COMMENTARY Ryanodine receptor cluster size sets the tone in cerebral smooth muscle COMMENTARY Christian Soellera,1 + Ryanodine receptors (RyRs) are large intracellular Ca2 WT mdx channels that provide the molecular basis of the pro- K+ K+ higher BK + + + cess termed Ca2 -induced Ca2 release (1). Ca2 sig- activity naling through RyRs has been shown to be critical for CaC BK BK skeletal, cardiac, and smooth muscle physiology (2) as well as for neurons (3) and secretory cells like pancre- larger RyR atic beta cells. RyRs were first seen in the electron RyRs RyRs clusters microscope as ∼30-nm-size particles (4), and it had SMCs been known from EM studies that they form clusters, SR SR typically in the narrow “junctional” space between sar- colemma and sarcoplasmic reticulum (SR) membranes. + Due to the inherent positive feedback of Ca2 -induced + Ca2 release, the size and shape of RyR clusters are expected to affect their functional activity. Indeed, de- SMCs SMCs tailed mathematical modeling suggests that RyR cluster- cerebral artery ing is important for determining the excitability of RyR- + mediated Ca2 release (5, 6) and could, in principle, also + affect the amount of Ca2 released in microscopic re- 2+ lease events termed Ca sparks (7). The experimental myogenic tone study of RyR clustering has received a boost with the reduced advent of optical superresolution microscopy, which myogenic has made the assessment of RyR cluster size more acces- sible by using essentially standard immunolabeling pro- tone tocols. When first applied in cardiac muscle cells, this myogenic tone (%) tone myogenic approach revealed a broad, approximately exponential vessel pressure (%) tone myogenic vessel pressure RyR cluster size distribution (8), which had previously not Fig. -

An Investigative Mathematical Model of Calcium-Induced Calcium Release
Biophysical Journal Volume 83 July 2002 59–78 59 Termination of Cardiac Ca2؉ Sparks: An Investigative Mathematical Model of Calcium-Induced Calcium Release Eric A. Sobie,* Keith W. Dilly,* Jader dos Santos Cruz,* W. Jonathan Lederer,* and M. Saleet Jafri† *Medical Biotechnology Center, University of Maryland Biotechnology Center, Baltimore, Maryland 21201, and †Department of Mathematical Sciences, The University of Texas at Dallas, Richardson, Texas 75083 USA ABSTRACT A Ca2ϩ spark arises when a cluster of sarcoplasmic reticulum (SR) channels (ryanodine receptors or RyRs) opens to release calcium in a locally regenerative manner. Normally triggered by Ca2ϩ influx across the sarcolemmal or transverse tubule membrane neighboring the cluster, the Ca2ϩ spark has been shown to be the elementary Ca2ϩ signaling event of excitation–contraction coupling in heart muscle. However, the question of how the Ca2ϩ spark terminates remains a central, unresolved issue. Here we present a new model, “sticky cluster,” of SR Ca2ϩ release that simulates Ca2ϩ spark behavior and enables robust Ca2ϩ spark termination. Two newly documented features of RyR behavior have been incorpo- rated in this otherwise simple model: “coupled gating” and an opening rate that depends on SR lumenal [Ca2ϩ]. Using a Monte Carlo method, local Ca2ϩ-induced Ca2ϩ release from clusters containing between 10 and 100 RyRs is modeled. After release is triggered, Ca2ϩ flux from RyRs diffuses into the cytosol and binds to intracellular buffers and the fluorescent Ca2ϩ indicator fluo-3 to produce the model Ca2ϩ spark. Ca2ϩ sparks generated by the sticky cluster model resemble those observed experimentally, and Ca2ϩ spark duration and amplitude are largely insensitive to the number of RyRs in a cluster.