CEBPD Amplification and Overexpression in Urothelial Carcinoma: a Driver of Tumor Metastasis Indicating Adverse Prognosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -

Open Chromatin Profiling in Adipose Tissue Marks Genomic Regions with Functional Roles in Cardiometabolic Traits
INVESTIGATION Open Chromatin Profiling in Adipose Tissue Marks Genomic Regions with Functional Roles in Cardiometabolic Traits Maren E. Cannon,*,1 Kevin W. Currin,*,1 Kristin L. Young,† Hannah J. Perrin,* Swarooparani Vadlamudi,* Alexias Safi,‡ Lingyun Song,‡ Ying Wu,* Martin Wabitsch,§ Markku Laakso,** Gregory E. Crawford,‡ and Karen L. Mohlke*,2 *Department of Genetics, †Department of Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, ‡Department of Pediatrics, Division of Medical Genetics and Center for Genomic and Computational Biology, § Duke University, NC 27710, Department of Pediatrics and Adolescents Medicine, Division of Pediatric Endocrinology and Diabetes, University of Ulm, Germany 89081, and **Department of Medicine, University of Eastern Finland and Kuopio University Hospital, Kuopio, Finland 70210 ORCID ID: 0000-0001-6721-153X (K.L.M.) ABSTRACT Identifying the regulatory mechanisms of genome-wide association study (GWAS) loci KEYWORDS affecting adipose tissue has been restricted due to limited characterization of adipose transcriptional chromatin regulatory elements. We profiled chromatin accessibility in three frozen human subcutaneous adipose accessibility tissue needle biopsies and preadipocytes and adipocytes from the Simpson Golabi-Behmel Syndrome adipose tissue (SGBS) cell strain using an assay for transposase-accessible chromatin (ATAC-seq). We identified preadipocytes 68,571 representative accessible chromatin regions (peaks) across adipose tissue samples (FDR , 5%). GWAS GWAS loci for eight cardiometabolic traits were enriched in these peaks (P , 0.005), with the strongest cardiometabolic enrichment for waist-hip ratio. Of 110 recently described cardiometabolic GWAS loci colocalized with trait adiposetissueeQTLs,59locihadoneormorevariantsoverlappinganadiposetissuepeak.Annotated variants at the SNX10 waist-hip ratio locus and the ATP2A1-SH2B1 body mass index locus showed allelic differences in regulatory assays. -

Transcription Factor Gene Expression Profiling and Analysis of SOX Gene Family Transcription Factors in Human Limbal Epithelial
Transcription factor gene expression profiling and analysis of SOX gene family transcription factors in human limbal epithelial progenitor cells Der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Dr. med. Johannes Menzel-Severing aus Bonn Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 7. Februar 2018 Vorsitzender des Promotionsorgans: Prof. Dr. Georg Kreimer Gutachter: Prof. Dr. Andreas Feigenspan Prof. Dr. Ursula Schlötzer-Schrehardt 1 INDEX 1. ABSTRACTS Page 1.1. Abstract in English 4 1.2. Zusammenfassung auf Deutsch 7 2. INTRODUCTION 2.1. Anatomy and histology of the cornea and the corneal surface 11 2.2. Homeostasis of corneal epithelium and the limbal stem cell paradigm 13 2.3. The limbal stem cell niche 15 2.4. Cell therapeutic strategies in ocular surface disease 17 2.5. Alternative cell sources for transplantation to the corneal surface 18 2.6. Transcription factors in cell differentiation and reprogramming 21 2.7. Transcription factors in limbal epithelial cells 22 2.8. Research question 25 3. MATERIALS AND METHODS 3.1. Human donor corneas 27 3.2. Laser Capture Microdissection (LCM) 28 3.3. RNA amplification and RT2 profiler PCR arrays 29 3.4. Real-time PCR analysis 33 3.5. Immunohistochemistry 34 3.6. Limbal epithelial cell culture 38 3.7. Transcription-factor knockdown/overexpression in vitro 39 3.8. Proliferation assay 40 3.9. Western blot 40 3.10. Statistical analysis 41 2 4. RESULTS 4.1. Quality control of LCM-isolated and amplified RNA 42 4.2. -

Xo GENE PANEL
xO GENE PANEL Targeted panel of 1714 genes | Tumor DNA Coverage: 500x | RNA reads: 50 million Onco-seq panel includes clinically relevant genes and a wide array of biologically relevant genes Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 -

In Vitro Targeting of Transcription Factors to Control the Cytokine Release Syndrome in 2 COVID-19 3
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.29.424728; this version posted December 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 In vitro Targeting of Transcription Factors to Control the Cytokine Release Syndrome in 2 COVID-19 3 4 Clarissa S. Santoso1, Zhaorong Li2, Jaice T. Rottenberg1, Xing Liu1, Vivian X. Shen1, Juan I. 5 Fuxman Bass1,2 6 7 1Department of Biology, Boston University, Boston, MA 02215, USA; 2Bioinformatics Program, 8 Boston University, Boston, MA 02215, USA 9 10 Corresponding author: 11 Juan I. Fuxman Bass 12 Boston University 13 5 Cummington Mall 14 Boston, MA 02215 15 Email: [email protected] 16 Phone: 617-353-2448 17 18 Classification: Biological Sciences 19 20 Keywords: COVID-19, cytokine release syndrome, cytokine storm, drug repurposing, 21 transcriptional regulators 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.12.29.424728; this version posted December 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 22 Abstract 23 Treatment of the cytokine release syndrome (CRS) has become an important part of rescuing 24 hospitalized COVID-19 patients. Here, we systematically explored the transcriptional regulators 25 of inflammatory cytokines involved in the COVID-19 CRS to identify candidate transcription 26 factors (TFs) for therapeutic targeting using approved drugs. -

Gene Regulatory Networks Underlying Human Microglia Maturation 2 3 Claudia Z
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.02.446636; this version posted June 2, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Gene regulatory networks underlying human microglia maturation 2 3 Claudia Z. Han1*, Rick Z. Li1*, Emily Hansen2,3, Hunter R. Bennett1 , Olivier Poirion4, Justin 4 Buchanan1,4, Jean F. Challacombe1, Bethany R. Fixsen1, Samantha Trescott2,3, Johannes C.M. 5 Schlachetzki1, Sebastian Preissl1,4, Allen Wang1,4, Carolyn O’Connor5, Anna S. Warden1,2,3, 6 Shreya Shriram2,3 , Roy Kim2,3, Celina T. Nguyen1,2,3, Danielle M. Schafer2,3, Gabriela Ramirez2,3, 7 Samuel A. Anavim2,3, Avalon Johnson2,3, Eniko Sajti2, Mihir Gupta6, Michael L. Levy6, Sharona 8 Ben-Haim6, David D. Gonda6, Louise Laurent7, Christopher K. Glass1, Nicole G. Coufal2,3,5 9 10 1 Department of Cellular and Molecular Medicine, University of California, San Diego, La Jolla, 11 CA 92093, USA. 12 2 Department of Pediatrics, University of California, San Diego, La Jolla, CA 92093 13 3 Sanford Consortium for Regenerative Medicine 14 4 Center for Epigenomics, University of California, San Diego, La Jolla, CA 92093 15 5 Salk Institute for Biological Studies, La Jolla, CA 92037 16 6 Department of Neurosurgery, University of California, San Diego-Rady Children's Hospital, San 17 Diego, CA 92123, USA 18 7 Department of Obstetrics, Gynecology, and Reproductive Sciences, University of California, 19 San Diego, CA 92123 20 *These authors contributed equally to this work. -

The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy
International Journal of Molecular Sciences Review The Multifaced Role of STAT3 in Cancer and Its Implication for Anticancer Therapy Manlio Tolomeo * and Antonio Cascio Department of Health Promotion Sciences, Maternal and Infant Care, Internal Medicine and Medical Specialties, University of Palermo, via del Vespro 129, 90127 Palermo, Italy; [email protected] * Correspondence: [email protected] Abstract: Signal transducer and activator of transcription (STAT) 3 is one of the most complex regulators of transcription. Constitutive activation of STAT3 has been reported in many types of tumors and depends on mechanisms such as hyperactivation of receptors for pro-oncogenic cytokines and growth factors, loss of negative regulation, and excessive cytokine stimulation. In contrast, somatic STAT3 mutations are less frequent in cancer. Several oncogenic targets of STAT3 have been recently identified such as c-myc, c-Jun, PLK-1, Pim1/2, Bcl-2, VEGF, bFGF, and Cten, and inhibitors of STAT3 have been developed for cancer prevention and treatment. However, despite the oncogenic role of STAT3 having been widely demonstrated, an increasing amount of data indicate that STAT3 functions are multifaced and not easy to classify. In fact, the specific cellular role of STAT3 seems to be determined by the integration of multiple signals, by the oncogenic environment, and by the alternative splicing into two distinct isoforms, STAT3α and STAT3β. On the basis of these different conditions, STAT3 can act both as a potent tumor promoter or tumor suppressor factor. This implies that the therapies based on STAT3 modulators should be performed considering the pleiotropic functions of this transcription factor and tailored to the specific tumor type. -
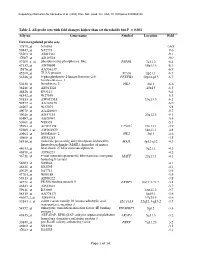
Table 2. All Probe Sets with Fold Changes Higher Than Set Thresholds but P > 0.001 Affy No
Supporting information for Sanoudou et al. (2003) Proc. Natl. Acad. Sci. USA, 10.1073/pnas.0330960100 Table 2. All probe sets with fold changes higher than set thresholds but P > 0.001 Affy no. Gene name Symbol Location Fold Down-regulated probe sets 47879_at N46863 -16.9 59447_at N52773 -9.6 55503_at AI085361 -9.1 47087_at AI310524 -9.0 37209_g_at phosphoserine phosphatase-like PSPHL 7q11.2 -8.4 47137_at AI479899 19p13.3 -8.1 45876_at AA536137 -6.9 45260_at TU3A protein TU3A 3p21.1 -6.7 56246_at 6-phosphofructo-2-kinase/fructose-2,6- PFKFB3 10p14-p15 -6.7 bisphosphatase 3 50230_at hexokinase 2 HK2 2p13 -6.6 38248_at AB011124 20p13 -6.5 44426_at R93141 -6.4 48542_at W27559 -6.3 53813_at AW051518 13q13.3 -6.1 59577_at AA243670 -6.0 46907_at W37075 -5.8 49078_at AA424983 -5.7 49026_at AI357153 20p12.3 -5.4 53487_at AI670947 -5.4 50161_at N39328 -5.1 35994_at AC002398 F25965 19q13.1 -4.9 52285_f_at AW002970 18p11.1 -4.8 40964_at hexokinase 2 HK2 2p13 -4.6 49806_at AI932283 -4.5 58918_at molecule possessing ankyrin repeats induced by MAIL 3p12-q12 -4.3 lipopolysaccharide (MAIL), homolog of mouse 44633_at heat shock 27 kDa associated protein 3q21.1 -4.3 46858_at AI796221 -4.2 36711_at v-maf musculoaponeurotic fibrosarcoma oncogene MAFF 22q13.1 -4.1 homolog F (avian) 54683_at N49844 -4.1 46621_at N32595 -4.1 49629_at N47713 -3.9 47703_at W89189 -3.9 59313_at AI598222 -3.8 34721_at FK506 binding protein 5 FKBP5 6p21.3-21.2 -3.8 46843_at AI632621 -3.7 59611_at R53069 16p11.2 -3.7 58315_at AA778171 3p25.1 -3.6 46607_f_at AI885018 17q25.3 -3.6 33143_s_at solute carrier family 16 (monocarboxylic acid SLC16A3 22q12.3-q13.2 -3.5 transporters), member 3 54152_at eukaryotic translation initiation factor 4E binding EIF4EBP1 8p12 -3.4 protein 1 43935_at ARF-GAP, RHO-GAP, ankyrin repeat and plekstrin ARAP3 5q31.3 -3.2 homology domains-containing protein 3 33849_at pre-B-cell colony-enhancing factor PBEF 7q11.23 -3.2 46902_at N92294 -3.2 47023_at N25555 -3.1 Page 1 of 14 Supporting information for Sanoudou et al. -

A Single-Cell Transcriptome Atlas of the Mouse Glomerulus
RAPID COMMUNICATION www.jasn.org A Single-Cell Transcriptome Atlas of the Mouse Glomerulus Nikos Karaiskos,1 Mahdieh Rahmatollahi,2 Anastasiya Boltengagen,1 Haiyue Liu,1 Martin Hoehne ,2 Markus Rinschen,2,3 Bernhard Schermer,2,4,5 Thomas Benzing,2,4,5 Nikolaus Rajewsky,1 Christine Kocks ,1 Martin Kann,2 and Roman-Ulrich Müller 2,4,5 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background Three different cell types constitute the glomerular filter: mesangial depending on cell location relative to the cells, endothelial cells, and podocytes. However, to what extent cellular heteroge- glomerular vascular pole.3 Because BP ad- neity exists within healthy glomerular cell populations remains unknown. aptation and mechanoadaptation of glo- merular cells are key determinants of kidney Methods We used nanodroplet-based highly parallel transcriptional profiling to function and dysregulated in kidney disease, characterize the cellular content of purified wild-type mouse glomeruli. we tested whether glomerular cell type sub- Results Unsupervised clustering of nearly 13,000 single-cell transcriptomes identi- sets can be identified by single-cell RNA fied the three known glomerular cell types. We provide a comprehensive online sequencing in wild-type glomeruli. This atlas of gene expression in glomerular cells that can be queried and visualized using technique allows for high-throughput tran- an interactive and freely available database. Novel marker genes for all glomerular scriptome profiling of individual cells and is cell types were identified and supported by immunohistochemistry images particularly suitable for identifying novel obtained from the Human Protein Atlas. -

Micro RNA-Based Regulation of Genomics and Transcriptomics of Inflammatory Cytokines in COVID-19
medRxiv preprint doi: https://doi.org/10.1101/2021.06.08.21258565; this version posted June 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Micro RNA-based regulation of genomics and transcriptomics of inflammatory cytokines in COVID-19 Manoj Khokhar1, Sojit Tomo1, Purvi Purohit*1 Department of Biochemistry, All India Institute of Medical Sciences, Jodhpur 342005, India *Corresponding author and address Dr Purvi Purohit Additional Professor Department of Biochemistry All India Institute of Medical Sciences, Basni Industrial Area, Phase-2 Jodhpur-342005, India. Tel: 09928388223 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. medRxiv preprint doi: https://doi.org/10.1101/2021.06.08.21258565; this version posted June 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Abstract: Background: Coronavirus disease 2019 is characterized by the elevation of a wide spectrum of inflammatory mediators, which are associated with poor disease outcomes. We aimed at an in-silico analysis of regulatory microRNA and their transcription factors (TF) for these inflammatory genes that may help to devise potential therapeutic strategies in the future. -

Human Transcription Factors Responsive to Initial Reprogramming Predominantly Undergo Legitimate Reprogramming During Fbroblast Conversion to Ipscs Ricardo R
www.nature.com/scientificreports OPEN Human transcription factors responsive to initial reprogramming predominantly undergo legitimate reprogramming during fbroblast conversion to iPSCs Ricardo R. Cevallos1, Yvonne J. K. Edwards1,2, John M. Parant3, Bradley K. Yoder2 & Kejin Hu1* The four transcription factors OCT4, SOX2, KLF4, and MYC (OSKM) together can convert human fbroblasts to induced pluripotent stem cells (iPSCs). It is, however, perplexing that they can do so only for a rare population of the starting cells with a long latency. Transcription factors (TFs) defne identities of both the starting fbroblasts and the end product, iPSCs, and are also of paramount importance for the reprogramming process. It is critical to upregulate or activate the iPSC-enriched TFs while downregulate or silence the fbroblast-enriched TFs. This report explores the initial TF responses to OSKM as the molecular underpinnings for both the potency aspects and the limitation sides of the OSKM reprogramming. The authors frst defned the TF reprogramome, i.e., the full complement of TFs to be reprogrammed. Most TFs were resistant to OSKM reprogramming at the initial stages, an observation consistent with the inefciency and long latency of iPSC reprogramming. Surprisingly, the current analyses also revealed that most of the TFs (at least 83 genes) that did respond to OSKM induction underwent legitimate reprogramming. The initial legitimate transcriptional responses of TFs to OSKM reprogramming were also observed in the reprogramming fbroblasts from a diferent individual. Such early biased legitimate reprogramming of the responsive TFs aligns well with the robustness aspect of the otherwise inefcient and stochastic OSKM reprogramming. OCT4, SOX2, KLF4, and MYC (collectively OSKM) can convert human fbroblasts into induced pluripotent stem cells (iPSCs), which are the man-made version of embryonic stem cells (ESCs)1–3. -

Single-Cell RNA Landscape of Intratumoral Heterogeneity and Immunosuppressive Microenvironment in Advanced Osteosarcoma
ARTICLE https://doi.org/10.1038/s41467-020-20059-6 OPEN Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma Yan Zhou1,11, Dong Yang2,11, Qingcheng Yang2,11, Xiaobin Lv 3,11, Wentao Huang4,11, Zhenhua Zhou5, Yaling Wang 1, Zhichang Zhang2, Ting Yuan2, Xiaomin Ding1, Lina Tang 1, Jianjun Zhang1, Junyi Yin1, Yujing Huang1, Wenxi Yu1, Yonggang Wang1, Chenliang Zhou1, Yang Su1, Aina He1, Yuanjue Sun1, Zan Shen1, ✉ ✉ ✉ ✉ Binzhi Qian 6, Wei Meng7,8, Jia Fei9, Yang Yao1 , Xinghua Pan 7,8 , Peizhan Chen 10 & Haiyan Hu1 1234567890():,; Osteosarcoma is the most frequent primary bone tumor with poor prognosis. Through RNA- sequencing of 100,987 individual cells from 7 primary, 2 recurrent, and 2 lung metastatic osteosarcoma lesions, 11 major cell clusters are identified based on unbiased clustering of gene expression profiles and canonical markers. The transcriptomic properties, regulators and dynamics of osteosarcoma malignant cells together with their tumor microenvironment particularly stromal and immune cells are characterized. The transdifferentiation of malignant osteoblastic cells from malignant chondroblastic cells is revealed by analyses of inferred copy-number variation and trajectory. A proinflammatory FABP4+ macrophages infiltration is noticed in lung metastatic osteosarcoma lesions. Lower osteoclasts infiltration is observed in chondroblastic, recurrent and lung metastatic osteosarcoma lesions compared to primary osteoblastic osteosarcoma lesions. Importantly, TIGIT blockade enhances the cytotoxicity effects of the primary CD3+ T cells with high proportion of TIGIT+ cells against osteo- sarcoma. These results present a single-cell atlas, explore intratumor heterogeneity, and provide potential therapeutic targets for osteosarcoma. 1 Oncology Department of Shanghai Jiao Tong University Affiliated Sixth People’s Hospital, Shanghai 200233, China.