Snareopathies: Diversity in Mechanisms and Symptoms
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cytogenomic SNP Microarray - Fetal ARUP Test Code 2002366 Maternal Contamination Study Fetal Spec Fetal Cells
Patient Report |FINAL Client: Example Client ABC123 Patient: Patient, Example 123 Test Drive Salt Lake City, UT 84108 DOB 2/13/1987 UNITED STATES Gender: Female Patient Identifiers: 01234567890ABCD, 012345 Physician: Doctor, Example Visit Number (FIN): 01234567890ABCD Collection Date: 00/00/0000 00:00 Cytogenomic SNP Microarray - Fetal ARUP test code 2002366 Maternal Contamination Study Fetal Spec Fetal Cells Single fetal genotype present; no maternal cells present. Fetal and maternal samples were tested using STR markers to rule out maternal cell contamination. This result has been reviewed and approved by Maternal Specimen Yes Cytogenomic SNP Microarray - Fetal Abnormal * (Ref Interval: Normal) Test Performed: Cytogenomic SNP Microarray- Fetal (ARRAY FE) Specimen Type: Direct (uncultured) villi Indication for Testing: Patient with 46,XX,t(4;13)(p16.3;q12) (Quest: EN935475D) ----------------------------------------------------------------- ----- RESULT SUMMARY Abnormal Microarray Result (Male) Unbalanced Translocation Involving Chromosomes 4 and 13 Classification: Pathogenic 4p Terminal Deletion (Wolf-Hirschhorn syndrome) Copy number change: 4p16.3p16.2 loss Size: 5.1 Mb 13q Proximal Region Deletion Copy number change: 13q11q12.12 loss Size: 6.1 Mb ----------------------------------------------------------------- ----- RESULT DESCRIPTION This analysis showed a terminal deletion (1 copy present) involving chromosome 4 within 4p16.3p16.2 and a proximal interstitial deletion (1 copy present) involving chromosome 13 within 13q11q12.12. This -

The Role of Molecular Scaffolds at the Active Zone in Synaptic Vesicle Distribution and Release Probability
The Role of Molecular Scaffolds at the Active Zone in Synaptic Vesicle Distribution and Release Probability DISSERTATION To obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry and Pharmacy Freie Universität Berlin Berlin, 2016 by Christina Beis (née Hollmann) This thesis was completed under the supervision of Prof. Dr. Stephan Sigrist from February 2011 to June 2016 at the Institute for Biology/ Genetics of Freie Universität Berlin, Germany. 1st reviewer: Prof. Dr. Stephan Sigrist 2nd reviewer: Prof. Dr. Hans-Joachim Pflüger Date of Defense: 21.11.2016 Statement of Authorship I hereby declare that the work presented in this thesis has been written independently and without inappropriate support. All sources of information are referenced. I hereby declare that this thesis has not been submitted, either in the same or in a different form, to this or any other university for a degree. ____________________________ Christina Beis Contents 1. Summary / Zusammenfassung .................................................................................................. 1 Summary....................................................................................................................................... 1 Zusammenfassung ........................................................................................................................ 3 2. Introduction .................................................................................................................................... -

Complexin-1 Is a Biomarker of Synucleinopathy
DMM Advance Online Articles. Posted 20 January 2017 as doi: 10.1242/dmm.028035 Access the most recent version at http://dmm.biologists.org/lookup/doi/10.1242/dmm.028035 BLOOD RNA BIOMARKERS IN PRODROMAL PARK4 AND REM SLEEP BEHAVIOR DISORDER SHOW ROLE OF COMPLEXIN-1 LOSS FOR RISK OF PARKINSON’S DISEASE Suna Lahut1,2, Suzana Gispert1, Özgür Ömür1,2, Candan Depboylu3, Kay Seidel4, Jorge Antolio Domínguez-Bautista1, Nadine Brehm1, Hülya Tireli5, Karl Hackmann6, Caroline Pirkevi2, Barbara Leube7, Vincent Ries3, Kerstin Reim8, Nils Brose8, Wilfred F. den Dunnen9, Madrid Johnson10, Zsuzsanna Wolf11, Marc Schindewolf12, Wiebke Schrempf13, Kathrin Reetz14, Peter Young15, David Vadasz3, Achilleas S. Frangakis10, Evelin Schröck6, Helmuth Steinmetz1, Marina Jendrach1, Udo Rüb4, Ayşe Nazlı Başak2¶, Wolfgang Oertel3¶, Georg Auburger1¶ 1Exp. Neurology, Goethe University Medical School, 60590 Frankfurt/Main, Germany; 2NDAL, Boğaziçi University, 34342 Istanbul, Turkey; 3Dept. of Neurology, Philipps University, Baldingerstrasse, 35043 Marburg, Germany; 4Dr. Senckenberg Chronomedical Institute, Goethe University, 60590 Frankfurt/Main, Germany; 5Dept. of Neurology, Haydarpaşa Numune Training and Research Hospital, 34668 Istanbul, Turkey; 6Institute for Clinical Genetics, Faculty of Medicine Carl Gustav Carus, TU Dresden, Fetscherstrasse 74, 01307 Dresden, Germany; 7Institute of Human Genetics, Heinrich Heine University, 40225 Düsseldorf, Germany; 8Dept. of Molecular Neurobiology and Center for the Molecular Physiology of the Brain, Max Planck Institute of Experimental Medicine, 37075 Göttingen, Germany; 9Dept. of Pathology and Medical Biology, Medical Center, University, 9700 RB Groningen, Netherlands; 10Institut für Biophysik and Frankfurt Institute for Molecular Life Sciences, Goethe University, 60438 Frankfurt/Main, Germany; 11Hemophilia Centre, Medical Clinic III, Institute of Transfusion Medicine and Immunhematology Goethe University, 60590 Frankfurt/Main, Germany; 12Dept. -

Pharmacological Mechanisms Underlying the Hepatoprotective Effects of Ecliptae Herba on Hepatocellular Carcinoma
Hindawi Evidence-Based Complementary and Alternative Medicine Volume 2021, Article ID 5591402, 17 pages https://doi.org/10.1155/2021/5591402 Research Article Pharmacological Mechanisms Underlying the Hepatoprotective Effects of Ecliptae herba on Hepatocellular Carcinoma Botao Pan ,1 Wenxiu Pan ,2 Zheng Lu ,3 and Chenglai Xia 1,4 1Affiliated Foshan Maternity & Child Healthcare Hospital, Southern Medical University, Foshan 528000, China 2Department of Laboratory, Fifth People’s Hospital of Foshan, Foshan 528000, China 3Wuzhou Maternal and Child Health-Care Hospital, Wuzhou 543000, China 4School of Pharmaceutical Sciences, Southern Medical University, Guangzhou 510515, China Correspondence should be addressed to Zheng Lu; [email protected] and Chenglai Xia; [email protected] Received 30 January 2021; Revised 31 May 2021; Accepted 3 July 2021; Published 16 July 2021 Academic Editor: Hongcai Shang Copyright © 2021 Botao Pan et al. -is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background. -e number of hepatocellular carcinoma (HCC) cases worldwide has increased significantly. As a traditional Chinese medicine (TCM) with a long history, Ecliptae herba (EH) has been widely used in HCC patients in China, but its hepatoprotective mechanism is still unclear. Methods. In this study, we applied a network pharmacology-based strategy and experimental ver- ification to systematically unravel the underlying mechanisms of EH against HCC. First, six active ingredients of EH were screened from the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP) by the ADME method. Subsequently, 52 potential targets of 6 active ingredients acting on HCC were screened from various databases, including TCMSP, DGIdb, SwissTargetPrediction, CTD, and GeneCards. -

Single-Cell RNA-Seq of Dopaminergic Neurons Informs Candidate Gene Selection for Sporadic
bioRxiv preprint doi: https://doi.org/10.1101/148049; this version posted August 31, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Single-cell RNA-seq of dopaminergic neurons informs candidate gene selection for sporadic 2 Parkinson's disease 3 4 Paul W. Hook1, Sarah A. McClymont1, Gabrielle H. Cannon1, William D. Law1, A. Jennifer 5 Morton2, Loyal A. Goff1,3*, Andrew S. McCallion1,4,5* 6 7 1McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of 8 Medicine, Baltimore, Maryland, United States of America 9 2Department of Physiology Development and Neuroscience, University of Cambridge, 10 Cambridge, United Kingdom 11 3Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, 12 Maryland, United States of America 13 4Department of Comparative and Molecular Pathobiology, Johns Hopkins University School of 14 Medicine, Baltimore, Maryland, United States of America 15 5Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland, 16 United States of America 17 *, To whom correspondence should be addressed: [email protected] and [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/148049; this version posted August 31, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. -

Inaugural Dissertation
Molecular Mechanisms that Regulate Neurotransmission and Establish Presynaptic Homeostasis at the Drosophila melanogaster Neuromuscular Junction Inaugural Dissertation to obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry and Pharmacy of Freie Universität Berlin by Anthony William McCarthy from Dublin, Ireland February 2020 The experimental part of this thesis was conducted from September 2015 to November 2019 under the supervision of Dr. Alexander Walter at the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) and at the CharitéCrossOver, Charité Campus Berlin Mitte. 1st Reviewer: Dr. Alexander Walter 2nd Reviewer: Prof. Dr. Stephan Sigrist Date of Defence: Statement of Authorship I hereby declare that I am the sole author of this thesis and that I have not used any sources or tools other than those quoted. Use of work by any other author is identified as such and appropriately referenced. Berlin, February 2020 Anthony William McCarthy Acknowledgments I would like to take the opportunity to thank all the people who have been important and made an impact on me during my time in Berlin. I would first like to thank my supervisor, Dr. Alexander Walter, for his guidance and supervision over the years. His passion for science and expert knowledge as well as approachable nature made for insightful conversation, projecting me forward with my projects. I would also like to thank Prof. Dr. Stephan Sigrist for his valuable input during thesis committee meetings and for acting as a reviewer. I am grateful to Prof. Dr. Volker Haucke, the members of his lab and all those at the FMP in Buch for the plentiful feedback I received following thesis committee meetings and progress presentations. -
Primepcr™Assay Validation Report
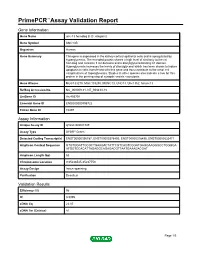
PrimePCR™Assay Validation Report Gene Information Gene Name unc-13 homolog B (C. elegans) Gene Symbol UNC13B Organism Human Gene Summary This gene is expressed in the kidney cortical epithelial cells and is upregulated by hyperglycemia. The encoded protein shares a high level of similarity to the rat homolog and contains 3 C2 domains and a diacylglycerol-binding C1 domain. Hyperglycemia increases the levels of diacylglycerol which has been shown to induce apoptosis in cells transfected with this gene and thus contribute to the renal cell complications of hyperglycemia. Studies in other species also indicate a role for this protein in the priming step of synaptic vesicle exocytosis. Gene Aliases MGC133279, MGC133280, MUNC13, UNC13, Unc13h2, hmunc13 RefSeq Accession No. NC_000009.11, NT_008413.18 UniGene ID Hs.493791 Ensembl Gene ID ENSG00000198722 Entrez Gene ID 10497 Assay Information Unique Assay ID qHsaCID0007349 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000396787, ENST00000378495, ENST00000378496, ENST00000535471 Amplicon Context Sequence GTGTGGATTGCGCTGAAGACTATTCGTCAGTCGGATGAGGAAGGGCCTGGGGA ATGGTCCACATTAGAGGCAGAGACGTTAATGAAAGACGAT Amplicon Length (bp) 63 Chromosome Location 9:35236545-35237753 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 96 R2 0.9995 cDNA Cq 22.07 cDNA Tm (Celsius) 81 Page 1/5 PrimePCR™Assay Validation Report gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report -

Meta-Analysis of Pharmacogenetic Interactions in Amyotrophic Lateral Sclerosis Clinical Trials
Published Ahead of Print on October 4, 2017 as 10.1212/WNL.0000000000004606 Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials Ruben P.A. van Eijk, MD ABSTRACT Ashley R. Jones, PhD Objective: To assess whether genetic subgroups in recent amyotrophic lateral sclerosis (ALS) tri- William Sproviero, PhD als responded to treatment with lithium carbonate, but that the treatment effect was lost in a large Aleksey Shatunov, PhD cohort of nonresponders. Pamela J. Shaw, MD, PhD Methods: Individual participant data were obtained from 3 randomized trials investigating the P. Nigel Leigh, MD, PhD efficacy of lithium carbonate. We matched clinical data with data regarding the UNC13A and Carolyn A. Young, MD, C9orf72 genotype. Our primary outcome was survival at 12 months. On an exploratory basis, we PhD assessed whether the effect of lithium depended on the genotype. Christopher E. Shaw, MD, PhD Results: Clinical data were available for 518 of the 606 participants. Overall, treatment with lith- Gabriele Mora, MD ium carbonate did not improve 12-month survival (hazard ratio [HR] 1.0, 95% confidence interval – p 5 UNC13A C9orf72 Jessica Mandrioli, MD [CI] 0.7 1.4; 0.96). Both the and genotype were independent predictors – p 5 – p 5 Giuseppe Borghero, MD of survival (HR 2.4, 95% CI 1.3 4.3; 0.006 and HR 2.5, 95% CI 1.1 5.2; 0.032, UNC13A p 5 Paolo Volanti, MD respectively). The effect of lithium was different for carriers ( 0.027), but not for C9orf72 p 5 UNC13A Frank P. Diekstra, MD, carriers ( 0.22). -

Reduced Insulin Secretion Correlates with Decreased Expression of Exocytotic Genes in Pancreatic Islets from Patients with Type 2 Diabetes
Molecular and Cellular Endocrinology 364 (2012) 36–45 Contents lists available at SciVerse ScienceDirect Molecular and Cellular Endocrinology journal homepage: www.elsevier.com/locate/mce Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes Sofia A. Andersson a, Anders H. Olsson b, Jonathan L.S. Esguerra a, Emilia Heimann e, Claes Ladenvall c, Anna Edlund a, Albert Salehi d, Jalal Taneera c, Eva Degerman e, Leif Groop c, Charlotte Ling b, ⇑ Lena Eliasson a, a Islet Cell Exocytosis, Lund University Diabetes Centre, Department of Clinical Sciences Malmö, Lund University, Malmö, Sweden b Epigenetics and Diabetes, Lund University Diabetes Centre, Department of Clinical Sciences Malmö, Lund University, Malmö, Sweden c Diabetes and Endocrinology, Lund University Diabetes Centre, Department of Clinical Sciences Malmö, Lund University, Malmö, Sweden d Islet Cell Physiology, Lund University Diabetes Centre, Department of Clinical Sciences Malmö, Lund University, Malmö, Sweden e Department of Experimental Medical Sciences, Biomedical Center, Lund University, Lund, Sweden article info abstract Article history: Reduced insulin release has been linked to defect exocytosis in b-cells. However, whether expression of Received 14 December 2011 genes suggested to be involved in the exocytotic process (exocytotic genes) is altered in pancreatic islets Received in revised form 7 August 2012 from patients with type 2 diabetes (T2D), and correlate to insulin secretion, needs to be further investi- Accepted 13 August 2012 gated. Available online 23 August 2012 Analysing expression levels of 23 exocytotic genes using microarray revealed reduced expression of five genes in human T2D islets (v2 = 13.25; p < 0.001). -

Genomic and Transcriptome Analysis Revealing an Oncogenic Functional Module in Meningiomas
Neurosurg Focus 35 (6):E3, 2013 ©AANS, 2013 Genomic and transcriptome analysis revealing an oncogenic functional module in meningiomas XIAO CHANG, PH.D.,1 LINGLING SHI, PH.D.,2 FAN GAO, PH.D.,1 JONATHAN RUssIN, M.D.,3 LIYUN ZENG, PH.D.,1 SHUHAN HE, B.S.,3 THOMAS C. CHEN, M.D.,3 STEVEN L. GIANNOTTA, M.D.,3 DANIEL J. WEISENBERGER, PH.D.,4 GAbrIEL ZADA, M.D.,3 KAI WANG, PH.D.,1,5,6 AND WIllIAM J. MAck, M.D.1,3 1Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, Los Angeles, California; 2GHM Institute of CNS Regeneration, Jinan University, Guangzhou, China; 3Department of Neurosurgery, Keck School of Medicine, University of Southern California, Los Angeles, California; 4USC Epigenome Center, Keck School of Medicine, University of Southern California, Los Angeles, California; 5Department of Psychiatry, Keck School of Medicine, University of Southern California, Los Angeles, California; and 6Division of Bioinformatics, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California Object. Meningiomas are among the most common primary adult brain tumors. Although typically benign, roughly 2%–5% display malignant pathological features. The key molecular pathways involved in malignant trans- formation remain to be determined. Methods. Illumina expression microarrays were used to assess gene expression levels, and Illumina single- nucleotide polymorphism arrays were used to identify copy number variants in benign, atypical, and malignant me- ningiomas (19 tumors, including 4 malignant ones). The authors also reanalyzed 2 expression data sets generated on Affymetrix microarrays (n = 68, including 6 malignant ones; n = 56, including 3 malignant ones). -

Whole-Genome Sequencing Reveals Genetic Variation in the Asian House Rat
INVESTIGATION Whole-Genome Sequencing Reveals Genetic Variation in the Asian House Rat Huajing Teng,*,†,‡ Yaohua Zhang,* Chengmin Shi,*,§ Fengbiao Mao,‡ Lingling Hou,‡ Hongling Guo,*,† Zhongsheng Sun,‡ and Jianxu Zhang*,1 *The State Key Laboratory of Integrated Management of Pest Insects and Rodents, Institute of Zoology, Chinese Academy of Sciences, 100101 Beijing, China, †University of Chinese Academy of Sciences, 100049 Beijing, China, § ‡ Beijing Institutes of Life Science, Chinese Academy of Sciences, 100101 Beijing, China, and Beijing Institute of Genomics, Chinese Academy of Sciences, 100101 Beijing, China ABSTRACT Whole-genome sequencing of wild-derived rat species can provide novel genomic resources, KEYWORDS which may help decipher the genetics underlying complex phenotypes. As a notorious pest, reservoir of human genetic pathogens, and colonizer, the Asian house rat, Rattus tanezumi, is successfully adapted to its habitat. However, landscape little is known regarding genetic variation in this species. In this study, we identified over 41,000,000 single- next-generation nucleotide polymorphisms, plus insertions and deletions, through whole-genome sequencing and bioinfor- sequencing matics analyses. Moreover, we identified over 12,000 structural variants, including 143 chromosomal inversions. Rattus tanezumi Further functional analyses revealed several fixed nonsense mutations associated with infection and immunity- single-nucleotide related adaptations, and a number of fixed missense mutations that may be related to anticoagulant resistance. polymorphisms A genome-wide scan for loci under selection identified various genes related to neural activity. Our whole- structural genome sequencing data provide a genomic resource for future genetic studies of the Asian house rat species variations and have the potential to facilitate understanding of the molecular adaptations of rats to their ecological niches. -

The Association of UNC13B Gene Polymorphisms and Diabetic
CLINICAL RESEARCH e-ISSN 1643-3750 © Med Sci Monit, 2019; 25: 8527-8533 DOI: 10.12659/MSM.919930 Received: 2019.09.05 Accepted: 2019.10.21 The Association of UNC13B Gene Polymorphisms Published: 2019.11.12 and Diabetic Kidney Disease in a Chinese Han Population Authors’ Contribution: AEG 1 Ya Wang 1 Department of Endocrinology, Jingzhou First People’s Hospital, The First Affiliated Study Design A AC 2 Jie Tan Hospital of Yangtze University, Jingzhou, Hubei, P.R. China Data Collection B 2 Department of Hematology, Jingzhou First People’s Hospital, The First Affiliated Statistical Analysis C BF 1 Dan Liu Hospital of Yangtze University, Jingzhou, Hubei, P.R .China Data Interpretation D BD 3 Yameng Yang 3 Department of Rheumatism and Immunology, Jingzhou First People’s Hospital, Manuscript Preparation E CG 1 Hongyan Wu The First Affiliated Hospital of Yangtze University, Jingzhou, Hubei, P.R. China Literature Search F Funds Collection G Corresponding Authors: Jie Tan, e-mail: [email protected], Hongyan Wu, e-mail: [email protected] Source of support: This study was supported by the Science and Technology Planning Project of Jingzhou Science and Technology Bureau, China (Grant No. 2018073) Background: Polymorphisms in the UNC13B gene are associated with diabetic kidney disease (DKD) in the European pop- ulation. Asian populations are more likely to suffer from complications of type 2 diabetes mellitus (T2DM), including diabetic kidney disease (DKD). This case-control study aimed to investigate the association between UNC13B gene polymorphisms and DKD in a Chinese Han population. Material/Methods: Five single nucleotide polymorphism (SNP) loci (rs13293564, rs17360668, rs10114937, rs661712, and rs2281999) were genotyped in the UNC13B gene in 600 Chinese Han subjects.