High-Throughput Drug Screens for Amyotrophic Lateral Sclerosis Drug Discovery
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Molecular Scaffolds at the Active Zone in Synaptic Vesicle Distribution and Release Probability
The Role of Molecular Scaffolds at the Active Zone in Synaptic Vesicle Distribution and Release Probability DISSERTATION To obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry and Pharmacy Freie Universität Berlin Berlin, 2016 by Christina Beis (née Hollmann) This thesis was completed under the supervision of Prof. Dr. Stephan Sigrist from February 2011 to June 2016 at the Institute for Biology/ Genetics of Freie Universität Berlin, Germany. 1st reviewer: Prof. Dr. Stephan Sigrist 2nd reviewer: Prof. Dr. Hans-Joachim Pflüger Date of Defense: 21.11.2016 Statement of Authorship I hereby declare that the work presented in this thesis has been written independently and without inappropriate support. All sources of information are referenced. I hereby declare that this thesis has not been submitted, either in the same or in a different form, to this or any other university for a degree. ____________________________ Christina Beis Contents 1. Summary / Zusammenfassung .................................................................................................. 1 Summary....................................................................................................................................... 1 Zusammenfassung ........................................................................................................................ 3 2. Introduction .................................................................................................................................... -

Inaugural Dissertation
Molecular Mechanisms that Regulate Neurotransmission and Establish Presynaptic Homeostasis at the Drosophila melanogaster Neuromuscular Junction Inaugural Dissertation to obtain the academic degree Doctor rerum naturalium (Dr. rer. nat.) submitted to the Department of Biology, Chemistry and Pharmacy of Freie Universität Berlin by Anthony William McCarthy from Dublin, Ireland February 2020 The experimental part of this thesis was conducted from September 2015 to November 2019 under the supervision of Dr. Alexander Walter at the Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) and at the CharitéCrossOver, Charité Campus Berlin Mitte. 1st Reviewer: Dr. Alexander Walter 2nd Reviewer: Prof. Dr. Stephan Sigrist Date of Defence: Statement of Authorship I hereby declare that I am the sole author of this thesis and that I have not used any sources or tools other than those quoted. Use of work by any other author is identified as such and appropriately referenced. Berlin, February 2020 Anthony William McCarthy Acknowledgments I would like to take the opportunity to thank all the people who have been important and made an impact on me during my time in Berlin. I would first like to thank my supervisor, Dr. Alexander Walter, for his guidance and supervision over the years. His passion for science and expert knowledge as well as approachable nature made for insightful conversation, projecting me forward with my projects. I would also like to thank Prof. Dr. Stephan Sigrist for his valuable input during thesis committee meetings and for acting as a reviewer. I am grateful to Prof. Dr. Volker Haucke, the members of his lab and all those at the FMP in Buch for the plentiful feedback I received following thesis committee meetings and progress presentations. -

Meta-Analysis of Pharmacogenetic Interactions in Amyotrophic Lateral Sclerosis Clinical Trials
Published Ahead of Print on October 4, 2017 as 10.1212/WNL.0000000000004606 Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials Ruben P.A. van Eijk, MD ABSTRACT Ashley R. Jones, PhD Objective: To assess whether genetic subgroups in recent amyotrophic lateral sclerosis (ALS) tri- William Sproviero, PhD als responded to treatment with lithium carbonate, but that the treatment effect was lost in a large Aleksey Shatunov, PhD cohort of nonresponders. Pamela J. Shaw, MD, PhD Methods: Individual participant data were obtained from 3 randomized trials investigating the P. Nigel Leigh, MD, PhD efficacy of lithium carbonate. We matched clinical data with data regarding the UNC13A and Carolyn A. Young, MD, C9orf72 genotype. Our primary outcome was survival at 12 months. On an exploratory basis, we PhD assessed whether the effect of lithium depended on the genotype. Christopher E. Shaw, MD, PhD Results: Clinical data were available for 518 of the 606 participants. Overall, treatment with lith- Gabriele Mora, MD ium carbonate did not improve 12-month survival (hazard ratio [HR] 1.0, 95% confidence interval – p 5 UNC13A C9orf72 Jessica Mandrioli, MD [CI] 0.7 1.4; 0.96). Both the and genotype were independent predictors – p 5 – p 5 Giuseppe Borghero, MD of survival (HR 2.4, 95% CI 1.3 4.3; 0.006 and HR 2.5, 95% CI 1.1 5.2; 0.032, UNC13A p 5 Paolo Volanti, MD respectively). The effect of lithium was different for carriers ( 0.027), but not for C9orf72 p 5 UNC13A Frank P. Diekstra, MD, carriers ( 0.22). -

Munc13-1/2 (T-20): Sc-13630
SAN TA C RUZ BI OTEC HNOL OG Y, INC . Munc13-1/2 (T-20): sc-13630 BACKGROUND PRODUCT Munc13 proteins (Munc13-1, Munc13-2, and Munc13-3) make up a family of Each vial contains 200 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide highly homologous synaptic molecules that bind Syntaxin, an essential medi - and 0.1% gelatin. ator of neurotransmitter release. Munc13 proteins contain phorbol ester bind - Blocking peptide available for competition studies, sc-13630 P, (100 µg ing C1- and C2-domains, which are regulatory domains for Ca 2+ , phospho - pep tide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). lipids and diacylglycerol. Munc13 proteins are primarily expressed by neu rons, except for a ubiquitously expressed Munc13-2 splice variant. Munc13-1 is APPLICATIONS expressed by most neurons; it interacts with the N-terminal of Doc2 α, which is concentrated on the synaptic vesicle. Munc13-1 also interacts directly with Munc13-1/2 (T-20) is recommended for detection of Munc13-1 and Munc13- 2 msec7-1 to co-localize the two proteins at the active zone, a presynaptic, of mouse, rat and human origin by Western Blotting (starting dilution 1:200, subcellular compartment with extremely high membrane turnover. Munc13-1 dilution range 1:100-1:1000), immunofluorescence (starting dilution 1:50, is essential for synaptic vesicle maturation and plays a role in the central dilution range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, priming function in synaptic vesicle exocytosis from glutamatergic synapses. -

Snareopathies: Diversity in Mechanisms and Symptoms
VU Research Portal SNAREopathies Verhage, Matthijs; Sørensen, Jakob B. published in Neuron 2020 DOI (link to publisher) 10.1016/j.neuron.2020.05.036 document version Publisher's PDF, also known as Version of record document license Article 25fa Dutch Copyright Act Link to publication in VU Research Portal citation for published version (APA) Verhage, M., & Sørensen, J. B. (2020). SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron, 107(1), 22-37. https://doi.org/10.1016/j.neuron.2020.05.036 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain • You may freely distribute the URL identifying the publication in the public portal ? Take down policy If you believe that this document breaches copyright please contact us providing details, and we will remove access to the work immediately and investigate your claim. E-mail address: [email protected] Download date: 29. Sep. 2021 ll Review SNAREopathies: Diversity in Mechanisms and Symptoms Matthijs Verhage1,2,* and Jakob B. Sørensen3,* 1Department of Functional Genomics, Vrije Universiteit (VU) Amsterdam, De Boelelaan 1085, Amsterdam 1081 HV, the Netherlands 2Department of Clinical Genetics, UMC Amsterdam, De Boelelaan 1085, Amsterdam 1081 HV, the Netherlands 3Department of Neuroscience, University of Copenhagen, 2200 Copenhagen N, Denmark *Correspondence: [email protected] (M.V.), [email protected] (J.B.S.) https://doi.org/10.1016/j.neuron.2020.05.036 Neuronal SNAREs and their key regulators together drive synaptic vesicle exocytosis and synaptic transmis- sion as a single integrated membrane fusion machine. -

GSE50161, (C) GSE66354, (D) GSE74195 and (E) GSE86574
Figure S1. Boxplots of normalized samples in five datasets. (A) GSE25604, (B) GSE50161, (C) GSE66354, (D) GSE74195 and (E) GSE86574. The x‑axes indicate samples, and the y‑axes represent the expression of genes. Figure S2. Volanco plots of DEGs in five datasets. (A) GSE25604, (B) GSE50161, (C) GSE66354, (D) GSE74195 and (E) GSE86574. Red nodes represent upregulated DEGs and green nodes indicate downregulated DEGs. Cut‑off criteria were P<0.05 and |log2 FC|>1. DEGs, differentially expressed genes; FC, fold change; adj.P.Val, adjusted P‑value. Figure S3. Transcription factor‑gene regulatory network constructed using the Cytoscape iRegulion plug‑in. Table SI. Primer sequences for reverse transcription‑quantitative polymerase chain reaction. Genes Sequences hsa‑miR‑124 F: 5'‑ACACTCCAGCTGGGCAGCAGCAATTCATGTTT‑3' R: 5'‑CTCAACTGGTGTCGTGGA‑3' hsa‑miR‑330‑3p F: 5'‑CATGAATTCACTCTCCCCGTTTCTCCCTCTGC‑3' R: 5'‑CCTGCGGCCGCGAGCCGCCCTGTTTGTCTGAG‑3' hsa‑miR‑34a‑5p F: 5'‑TGGCAGTGTCTTAGCTGGTTGT‑3' R: 5'‑GCGAGCACAGAATTAATACGAC‑3' hsa‑miR‑449a F: 5'‑TGCGGTGGCAGTGTATTGTTAGC‑3' R: 5'‑CCAGTGCAGGGTCCGAGGT‑3' CD44 F: 5'‑CGGACACCATGGACAAGTTT‑3' R: 5'‑TGTCAATCCAGTTTCAGCATCA‑3' PCNA F: 5'‑GAACTGGTTCATTCATCTCTATGG‑3' F: 5'‑TGTCACAGACAAGTAATGTCGATAAA‑3' SYT1 F: 5'‑CAATAGCCATAGTCGCAGTCCT‑3' R: 5'‑TGTCAATCCAGTTTCAGCATCA‑3' U6 F: 5'‑GCTTCGGCAGCACATATACTAAAAT‑3' R: 5'‑CGCTTCACGAATTTGCGTGTCAT‑3' GAPDH F: 5'‑GGAAAGCTGTGGCGTGAT‑3' R: 5'‑AAGGTGGAAGAATGGGAGTT‑3' hsa, homo sapiens; miR, microRNA; CD44, CD44 molecule (Indian blood group); PCNA, proliferating cell nuclear antigen; -

Soluble Α-Synuclein Facilitates Priming and Fusion by Releasing Ca2+ From
© 2018. Published by The Company of Biologists Ltd | Journal of Cell Science (2018) 131, jcs213017. doi:10.1242/jcs.213017 RESEARCH ARTICLE Soluble α-synuclein facilitates priming and fusion by releasing Ca2+ from the thapsigargin-sensitive Ca2+ pool in PC12 cells Chien-Chang Huang1,2, Tai-Yu Chiu2, Tzu-Ying Lee2, Hsin-Jui Hsieh2, Chung-Chih Lin1,2,3,* and Lung-Sen Kao1,2,* ABSTRACT abnormal aggregation and neuronal degeneration both at the cellular α-Synuclein is associated with Parkinson’s disease, and is mainly level (Outeiro et al., 2008) and in a mouse model (Masliah et al., α localized in presynaptic terminals and regulates exocytosis, but its 2000). Studies on transgenic mice overexpressing -synuclein, α physiological roles remain controversial. Here, we studied the effects show that -synuclein has at least three distinct subcellular forms in of soluble and aggregated α-synuclein on exocytosis, and explored presynaptic terminals. These consist of unbound soluble protein, the molecular mechanism by which α-synuclein interacts with proteins bound to presynaptic vesicles and proteins in the form of regulatory proteins, including Rab3A, Munc13-1 (also known as microaggregates (Spinelli et al., 2014). α Unc13a) and Munc18-1 (also known as STXBP1), in order to regulate Several lines of evidence have shown that -synuclein is involved α exocytosis. Through fluorescence recovery after photobleaching in the regulation of exocytosis. Firstly, -synuclein is mainly experiments, overexpressed α-synuclein in PC12 cells was found to localized within the presynaptic terminals and has been reported to be in a monomeric form, which promotes exocytosis. In contrast, regulate the size of the presynaptic vesicular pool, vesicular aggregated α-synuclein induced by lactacystin treatment inhibits turnover, synaptic plasticity and the assembly of the soluble NSF exocytosis. -

Bioinformatics Analysis of the Molecular Mechanisms Underlying Traumatic Spinal Cord Injury
8484 MOLECULAR MEDICINE REPORTS 17: 8484-8492, 2018 Bioinformatics analysis of the molecular mechanisms underlying traumatic spinal cord injury SHU-JIE ZHAO, WEI ZHOU, JIAN CHEN, YONG-JUN LUO and GUO-YONG YIN Department of Orthopedics, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu 210029, P.R. China Received October 17, 2017; Accepted February 7, 2018 DOI: 10.3892/mmr.2018.8918 Abstract. Spinal cord injury (SCI) is a cause of disability. The which is primarily caused by traffic accidents, occurs when an present study aimed to investigate the molecular mechanisms external impact acutely damages the spinal cord, and may be involved in traumatic SCI. Transcriptome data under acces- temporally divided into four stages: i) Acute at <48 h following sion no. GSE5296, including 96 chips, were downloaded from injury; ii) subacute at 48 h-14 days following injury; iii) inter- the Gene Expression Omnibus database. The raw data were mediate at 14 days-6 months following injury; and iv) chronic normalized and differentially expressed genes (DEGs) were at >6 months following injury (1). Although numerous studies identified. Furthermore, Kyoto Encyclopedia of Genes and have focused on neuroprotective and neuroregenerative Genomes pathway and Gene Ontology enrichment analysis of therapies, no major breakthroughs have been achieved (3-5). up- and downregulated DEGs was performed. Additionally, Therefore, it is necessary to elucidate the underlying mecha- a protein-protein interaction network was constructed and nism of SCI. the expression patterns of different genes were determined. Pathophysiologically, the initial mechanical injury damages Compared with sham samples, there were 374, 707, 1,322, neurons and initiates a complex secondary injury cascade, 1,475, 1,724 and 1,342 DEGs identified at 0.5, 4, 24 and 72 h, leading to progressive cell death, ischemia and inflamma- and 7 and 28 days post-injury, respectively. -
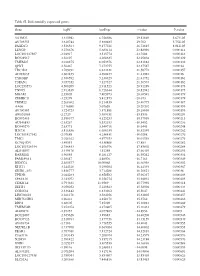
Table SI. Differentially Expressed Genes. Gene Logfc Aveexpr T Value
Table SI. Differentially expressed genes. Gene logFC AveExpr t value P‑value VSTM2L 3.315942 6.756386 29.82869 5.67E‑05 AV703555 3.610744 5.880485 29.702 5.75E‑05 FAXDC2 3.526564 9.177221 26.72815 8.01E‑05 LENG9 3.270676 5.683114 23.88996 0.000114 LOC101927507 ‑2.94917 3.515774 ‑23.7604 0.000116 BC028967 3.56197 4.208635 22.95804 0.000129 TMEM27 3.010875 8.053376 22.81882 0.000132 QPRT 2.56467 7.272755 22.37587 0.00014 TBC1D2 3.789191 6.823331 21.58772 0.000157 AF085835 2.602455 4.690477 21.43982 0.00016 TYROBP 2.386592 5.236329 21.41752 0.000161 TSPAN1 3.057352 7.127327 21.36574 0.000162 LOC253573 4.500209 2.911523 20.91289 0.000173 TNNC1 2.913839 6.726644 20.82942 0.000175 MGAM 2.35805 7.405973 20.68591 0.000179 ZDHHC11 ‑3.25279 5.412875 ‑20.673 0.000179 TRIM22 2.268602 8.214839 20.40775 0.000187 AA06 2.716698 5.07658 20.20202 0.000193 AV700385 3.724723 5.871881 20.18696 0.000193 AW851069 ‑2.2729 5.389451 ‑19.8536 0.000204 BC002465 2.590377 4.125253 19.57939 0.000213 AU146893 ‑2.63267 5.532153 ‑18.9452 0.000236 BC040270 ‑2.63771 5.540716 ‑18.6441 0.000248 H28731 3.816856 6.686159 18.32359 0.000262 LOC101927342 ‑2.07689 4.258431 ‑18.0208 0.000276 TMC1 2.028162 4.977353 18.01518 0.000276 KCNQ1DN ‑3.49551 4.538808 ‑17.883 0.000282 LOC101926934 2.786453 4.106976 17.85802 0.000284 AL043897 3.449478 4.51258 17.66439 0.000293 RARRES1 1.96055 8.281127 16.95242 0.000334 FAM189A1 ‑2.38147 4.48506 ‑16.7163 0.000349 IGDCC4 2.385577 10.00068 16.66958 0.000352 KLK11 3.244328 5.290104 16.44339 0.000367 GRID1‑AS1 1.858777 5.714288 16.26821 0.00038 UPK3B -

A Network View on Parkinson's Disease
Volume No: 7, Issue: 8, April 2013, e201304004, http://dx.doi.org/10.5936/csbj.201304004 CSBJ A Network View on Parkinson’s Disease Sreedevi Chandrasekaran a, Danail Bonchev a,* Abstract: Network-based systems biology tools including Pathway Studio 9.0 were used to identify Parkinson’s disease (PD) critical molecular players, drug targets, and underlying biological processes. Utilizing several microarray gene expression datasets, biomolecular networks such as direct interaction, shortest path, and microRNA regulatory networks were constructed and analyzed for the disease conditions. Network topology analysis of node connectivity and centrality revealed in combination with the guilt- by-association rule 17 novel genes of PD-potential interest. Seven new microRNAs (miR-132, miR-133a1, miR-181-1, miR-182, miR-218-1, miR-29a, and miR-330) related to Parkinson’s disease were identified, along with more microRNA targeted genes of interest like RIMS3, SEMA6D and SYNJ1. David and IPA enrichment analysis of KEGG and canonical pathways provided valuable mechanistic information emphasizing among others the role of chemokine signaling, adherence junction, and regulation of actin cytoskeleton pathways. Several routes for possible disease initiation and neuro protection mechanisms triggered via the extra- cellular ligands such as CX3CL1, SEMA6D and IL12B were thus uncovered, and a dual regulatory system of integrated transcription factors and microRNAs mechanisms was detected. 1. Introduction James Parkinson has been the first to observe this disease in adults including ATP13A2, DJ-1, GIGYF2, HTRA2, LRRK2, PARK2 in the year 1817. In his essay entitled “An Essay of the Shaking (parkin), PINK1, SNCA and UCHL1 were associated with either Palsy” he described this disease as initiated with slow, progressive autosomal dominant or recessive form of Parkinson’s disease [5]. -

Distinct Genetic Signatures of Cortical and Subcortical Regions Associated with Human Memory
New Research Cognition and Behavior Distinct Genetic Signatures of Cortical and Subcortical Regions Associated with Human Memory Pin Kwang Tan,1 Egor Ananyev,2 and Po-Jang Hsieh3 https://doi.org/10.1523/ENEURO.0283-19.2019 1The N.1 Institute of Health, National University of Singapore, Singapore, Singapore 117456, 2Department of Psychology, Nanyang Technological University, Singapore, Singapore 639798, and 3Department of Psychology, National Taiwan University, Taipei City, Taiwan 10617 Abstract Despite the discovery of gene variants linked to memory performance, understanding the genetic basis of adult human memory remains a challenge. Here, we devised an unsupervised framework that relies on spatial correlations between human transcriptome data and functional neuroimaging maps to uncover the genetic signatures of memory in functionally-defined cortical and subcortical memory regions. Results were validated with animal literature and showed that our framework is highly effective in identifying memory-related processes and genes compared to a control cognitive function. Genes preferentially expressed in cortical memory regions are linked to memory-related processes such as immune and epigenetic regulation. Genes expressed in subcortical memory regions are associated with neurogenesis and glial cell differentiation. Genes expressed in both cortical and subcortical memory areas are involved in the regulation of transcription, synaptic plasticity, and glutamate receptor signaling. Furthermore, distinct memory- associated genes such as PRKCD and CDK5 are linked to cortical and subcortical regions, respectively. Thus, cortical and subcortical memory regions exhibit distinct genetic signatures that potentially reflect functional differences in health and disease, and nominates gene candidates for future experimental investigations. Key words: cognition; cortical; genetic; human; memory; neuroimaging Significance Statement The anatomic and functional aspects of human memory are well characterized, but its biological mecha- nisms are poorly understood. -

Synaptic UNC13A Protein Variant Causes Increased Neurotransmission and Dyskinetic Movement Disorder
Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder Noa Lipstein, … , Judith J. Jans, Nils Brose J Clin Invest. 2017;127(3):1005-1018. https://doi.org/10.1172/JCI90259. Research Article Neuroscience Munc13 proteins are essential regulators of neurotransmitter release at nerve cell synapses. They mediate the priming step that renders synaptic vesicles fusion-competent, and their genetic elimination causes a complete block of synaptic transmission. Here we have described a patient displaying a disorder characterized by a dyskinetic movement disorder, developmental delay, and autism. Using whole-exome sequencing, we have shown that this condition is associated with a rare, de novo Pro814Leu variant in the major human Munc13 paralog UNC13A (also known as Munc13-1). Electrophysiological studies in murine neuronal cultures and functional analyses in Caenorhabditis elegans revealed that the UNC13A variant causes a distinct dominant gain of function that is characterized by increased fusion propensity of synaptic vesicles, which leads to increased initial synaptic vesicle release probability and abnormal short-term synaptic plasticity. Our study underscores the critical importance of fine-tuned presynaptic control in normal brain function. Further, it adds the neuronal Munc13 proteins and the synaptic vesicle priming process that they control to the known etiological mechanisms of psychiatric and neurological synaptopathies. Find the latest version: https://jci.me/90259/pdf The Journal of Clinical Investigation RESEARCH ARTICLE Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder Noa Lipstein,1 Nanda M. Verhoeven-Duif,2,3 Francesco E. Michelassi,4 Nathaniel Calloway,4 Peter M. van Hasselt,5 Katarzyna Pienkowska,1 Gijs van Haaften,2,3 Mieke M.