Trajectory and Uniqueness of Mutational Signatures in Yeast Mutators
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Mutational Signatures: Emerging Concepts, Caveats and Clinical Applications
REVIEWS Mutational signatures: emerging concepts, caveats and clinical applications Gene Koh 1,2, Andrea Degasperi 1,2, Xueqing Zou1,2, Sophie Momen1,2 and Serena Nik-Zainal 1,2 ✉ Abstract | Whole-genome sequencing has brought the cancer genomics community into new territory. Thanks to the sheer power provided by the thousands of mutations present in each patient’s cancer, we have been able to discern generic patterns of mutations, termed ‘mutational signatures’, that arise during tumorigenesis. These mutational signatures provide new insights into the causes of individual cancers, revealing both endogenous and exogenous factors that have influenced cancer development. This Review brings readers up to date in a field that is expanding in computational, experimental and clinical directions. We focus on recent conceptual advances, underscoring some of the caveats associated with using the mutational signature frameworks and highlighting the latest experimental insights. We conclude by bringing attention to areas that are likely to see advancements in clinical applications. The concept of mutational signatures was introduced or drug therapies, in an attempt to decipher causes7–9. in 2012 following the demonstration that analy However, the origins of many signatures remain cryp sis of all substitution mutations in a set of 21 whole tic. Furthermore, while earlier analyses reported single genomesequenced (WGS) breast cancers could reveal signatures, for example of UV radiation (that is, SBS7)2, consistent patterns of mutagenesis across tumours1 more recent studies reported multiple versions of these (FIG. 1a). These patterns were the physiological imprints signatures (that is, SBS7a, SBS7b, SBS7c and SBS7d)3, of DNA damage and repair processes that had occurred leading the community to question whether some find during tumorigenesis and could distinguish BRCA1null ings are reflective of biology or are simply mathemat and BRCA2null tumours from sporadic breast cancers. -

3256.Full.Pdf
Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair Julie Akiko Heck, Juan Lucas Argueso, Zekeriyya Gemici, Richard Guy Reeves*, Ann Bernard, Charles F. Aquadro, and Eric Alani† Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853 Communicated by Thomas D. Petes, University of North Carolina, Chapel Hill, NC, December 20, 2005 (received for review June 22, 2005) In budding yeast, the MLH1-PMS1 heterodimer is the major MutL The influences of epistatic interactions on a wide variety of homolog complex that acts to repair mismatches arising during traits and processes have garnered increasing attention (14–17). DNA replication. Using a highly sensitive mutator assay, we ob- Few examples, however, have been characterized in molecular served that Saccharomyces cerevisiae strains bearing the S288c- detail. Here we show that the strain-dependent MMR pheno- strain-derived MLH1 gene and the SK1-strain-derived PMS1 gene types observed previously for site-specific mlh1 mutants were displayed elevated mutation rates that conferred a long-term due in part to an underlying defect between wild-type MMR fitness cost. Dissection of this negative epistatic interaction using genes from S288c and SK1. We identified the specific amino acid S288c-SK1 chimeras revealed that a single amino acid polymor- polymorphisms in MLH1 and PMS1, whose combined effect in phism in each gene accounts for this mismatch repair defect. Were hybrid strains leads to an elevation in mutation rate and a these strains to cross in natural populations, segregation of alleles generalized reduction in long-term fitness. -

DF6906-PMS1 Antibody
Affinity Biosciences website:www.affbiotech.com order:[email protected] PMS1 Antibody Cat.#: DF6906 Concn.: 1mg/ml Mol.Wt.: 106kDa Size: 50ul,100ul,200ul Source: Rabbit Clonality: Polyclonal Application: WB 1:500-1:2000, IHC 1:50-1:200, ELISA(peptide) 1:20000-1:40000 *The optimal dilutions should be determined by the end user. Reactivity: Human,Rat Purification: The antiserum was purified by peptide affinity chromatography using SulfoLink™ Coupling Resin (Thermo Fisher Scientific). Specificity: PMS1 Antibody detects endogenous levels of total PMS1. Immunogen: A synthesized peptide derived from human PMS1, corresponding to a region within the internal amino acids. Uniprot: P54277 Description: PMS1 belongs to the DNA mismatch repair mutL/hexB family. It is thought to be involved in the repair of DNA mismatches, and it can form heterodimers with MLH1, a known DNA mismatch repair protein. Mutations in PMS1 cause hereditary nonpolyposis colorectal cancer type 3 (HNPCC3) either alone or in combination with mutations in other proteins involved in the HNPCC phenotype, which is also known as Lynch syndrome (1). Storage Condition and Rabbit IgG in phosphate buffered saline , pH 7.4, 150mM Buffer: NaCl, 0.02% sodium azide and 50% glycerol.Store at -20 °C.Stable for 12 months from date of receipt. Western blot analysis of Hela whole cell lysates, using PMS1 Antibody. The lane on the left was treated with the antigen- specific peptide. 1 / 2 Affinity Biosciences website:www.affbiotech.com order:[email protected] DF6906 at 1/100 staining Human esophageal cancer by IHC-P. The sample was formaldehyde fixed and a heat mediated antigen retrieval step in citrate buffer was performed. -

Negative Epistasis Between Natural Variants of the Saccharomyces Cerevisiae MLH1 and PMS1 Genes Results in a Defect in Mismatch Repair
Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair Julie Akiko Heck, Juan Lucas Argueso, Zekeriyya Gemici, Richard Guy Reeves*, Ann Bernard, Charles F. Aquadro, and Eric Alani† Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853 Communicated by Thomas D. Petes, University of North Carolina, Chapel Hill, NC, December 20, 2005 (received for review June 22, 2005) In budding yeast, the MLH1-PMS1 heterodimer is the major MutL The influences of epistatic interactions on a wide variety of homolog complex that acts to repair mismatches arising during traits and processes have garnered increasing attention (14–17). DNA replication. Using a highly sensitive mutator assay, we ob- Few examples, however, have been characterized in molecular served that Saccharomyces cerevisiae strains bearing the S288c- detail. Here we show that the strain-dependent MMR pheno- strain-derived MLH1 gene and the SK1-strain-derived PMS1 gene types observed previously for site-specific mlh1 mutants were displayed elevated mutation rates that conferred a long-term due in part to an underlying defect between wild-type MMR fitness cost. Dissection of this negative epistatic interaction using genes from S288c and SK1. We identified the specific amino acid S288c-SK1 chimeras revealed that a single amino acid polymor- polymorphisms in MLH1 and PMS1, whose combined effect in phism in each gene accounts for this mismatch repair defect. Were hybrid strains leads to an elevation in mutation rate and a these strains to cross in natural populations, segregation of alleles generalized reduction in long-term fitness. -

The Repertoire of Mutational Signatures in Human Cancer
Article The repertoire of mutational signatures in human cancer https://doi.org/10.1038/s41586-020-1943-3 Ludmil B. Alexandrov1,25, Jaegil Kim2,25, Nicholas J. Haradhvala2,3,25, Mi Ni Huang4,5,25, Alvin Wei Tian Ng4,5, Yang Wu4,5, Arnoud Boot4,5, Kyle R. Covington6,7, Dmitry A. Gordenin8, Received: 18 May 2018 Erik N. Bergstrom1, S. M. Ashiqul Islam1, Nuria Lopez-Bigas9,10,11, Leszek J. Klimczak12, Accepted: 18 November 2019 John R. McPherson4,5, Sandro Morganella13, Radhakrishnan Sabarinathan10,14,15, David A. Wheeler6,16, Ville Mustonen17,18,19, PCAWG Mutational Signatures Working Group20, Published online: 5 February 2020 Gad Getz2,3,21,22,26, Steven G. Rozen4,5,23,26*, Michael R. Stratton13,26* & PCAWG Consortium24 The number of DBSs is proportional to the number of SBSs, with few exceptions Open access Somatic mutations in cancer genomes are caused by multiple mutational processes, each of which generates a characteristic mutational signature1. Here, as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium2 of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA), we characterized mutational signatures using 84,729,690 somatic mutations from 4,645 whole-genome and 19,184 exome sequences that encompass most types of cancer. We identifed 49 single-base-substitution, 11 doublet-base-substitution, 4 clustered-base-substitution and 17 small insertion-and-deletion signatures. The substantial size of our dataset, compared with previous analyses3–15, enabled the discovery of new signatures, the separation of overlapping signatures and the decomposition of signatures into components that may represent associated—but distinct—DNA damage, repair and/or replication mechanisms. -

Mutational Landscape in Genetically Engineered, Carcinogen- Induced, and Radiation-Induced Mouse Sarcoma
Mutational landscape in genetically engineered, carcinogen- induced, and radiation-induced mouse sarcoma Chang-Lung Lee, … , Kouros Owzar, David G. Kirsch JCI Insight. 2019. https://doi.org/10.1172/jci.insight.128698. Research In-Press Preview Genetics Oncology Cancer development is influenced by hereditary mutations, somatic mutations due to random errors in DNA replication, or external factors. It remains unclear how distinct cell-intrinsic and -extrinsic factors impact oncogenesis within the same tissue type. We investigated murine soft tissue sarcomas generated by oncogenic alterations (KrasG12D activation and p53 deletion), carcinogens (3-methylcholanthrene [MCA] or ionizing radiation), and in a novel model combining both factors (MCA plus p53 deletion). Whole-exome sequencing demonstrated distinct mutational signatures in individual sarcoma cohorts. MCA-induced sarcomas exhibited high mutational burden and predominantly G-to-T transversions, while radiation-induced sarcomas exhibited low mutational burden and a distinct genetic signature characterized by C-to-T transitions. The indel to substitution ratio and amount of gene copy number variations were high for radiation-induced sarcomas. MCA-induced tumors generated on a p53-deficient background showed the highest genomic instability. MCA- induced sarcomas harbored mutations in putative cancer-driver genes that regulate MAPK signaling (Kras and Nf1) and the Hippo pathway (Fat1 and Fat4). In contrast, radiation-induced sarcomas and KrasG12Dp53–/– sarcomas did not harbor recurrent oncogenic mutations, rather they exhibited amplifications of specific oncogenes: Kras and Myc in KrasG12Dp53–/– sarcomas, and Met and Yap1 for radiation-induced sarcomas. These results reveal that different initiating events drive oncogenesis through distinct mechanisms. Find the latest version: https://jci.me/128698/pdf Mutational landscape in genetically engineered, carcinogen-induced, and radiation-induced mouse sarcoma Chang-Lung Lee1,2,3*, Yvonne M. -

Clinical Outcome-Related Mutational Signatures Identified by Integrative
Author Manuscript Published OnlineFirst on September 28, 2020; DOI: 10.1158/1078-0432.CCR-20-2854 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Clinical Outcome-Related Mutational Signatures Identified by Integrative Genomic Analysis in Nasopharyngeal Carcinoma Wei Dai1,2*, Dittman Lai-Shun Chung1, Larry Ka-Yue Chow1, Valen Zhuoyou Yu1, Lisa Chan Lei1, Merrin Man-Long Leong1, Candy King-Chi Chan1, Josephine Mun-Yee Ko1, Maria Li Lung1* 1Department of Clinical Oncology, University of Hong Kong, Hong Kong (SAR), P. R. China 2University of Hong Kong-Shenzhen Hospital, Shenzhen, P. R. China Running title: Clinical Outcome-Related Mutational Signatures in NPC *Co-corresponding authors: Maria Li Lung, Wei Dai MLL Address: Department of Clinical Oncology, The University of Hong Kong, Room L6- 43, 6/F, Laboratory Block, Faculty of Medicine Building, 21 Sassoon Road, Pokfulam, Hong Kong Email: [email protected] Tel: (852) 3917 9783 Fax: (852) 2816 6279 WD Address: Department of Clinical Oncology, The University of Hong Kong, Room L10- 56, 10/F, Laboratory Block, Faculty of Medicine Building, 21 Sassoon Road, Pokfulam, Hong Kong Email: [email protected] Tel: (852) 3917 6930 Fax: (852) 2816 6279 Conflict of interest The authors declare no potential conflicts of interest Translational relevance In NPC, general consensus for treatment is to use radiotherapy (RT) alone for stage I disease, RT with or without concurrent chemotherapy for stage II and chemoradiotherapy (CRT) for advanced stage disease. However, 15-58% of the cases do not respond well to conventional treatment, and, thus, have poor clinical outcomes. -

PMS2 Antibody / PMS1 Homolog 2 (RQ4366)
PMS2 Antibody / PMS1 homolog 2 (RQ4366) Catalog No. Formulation Size RQ4366 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Antigen affinity purified Buffer Lyophilized from 1X PBS with 2% Trehalose and 0.025% sodium azide UniProt P54278 Localization Nucleus Applications Western blot : 0.5-1ug/ml Direct ELISA : 0.1-0.5ug/ml Limitations This PMS2 antibody is available for research use only. Western blot testing of human HeLa cell lysate with BCMA antibody at 0.5ug/ml. Expected molecular weight: 96-110 kDa. Description Mismatch repair endonuclease PMS2 is an enzyme that in humans is encoded by the PMS2 gene. The protein encoded by this gene is a key component of the mismatch repair system that functions to correct DNA mismatches and small insertions and deletions that can occur during DNA replication and homologous recombination. This protein forms heterodimers with the gene product of the mutL homolog 1 (MLH1) gene to form the MutL-alpha heterodimer. The MutL-alpha heterodimer possesses an endonucleolytic activity that is activated following recognition of mismatches and insertion/deletion loops by the MutS-alpha and MutS-beta heterodimers, and is necessary for removal of the mismatched DNA. There is a DQHA(X)2E(X)4E motif found at the C-terminus of the protein encoded by this gene that forms part of the active site of the nuclease. Mutations in this gene have been associated with hereditary nonpolyposis colorectal cancer (HNPCC; also known as Lynch syndrome) and Turcot syndrome. -
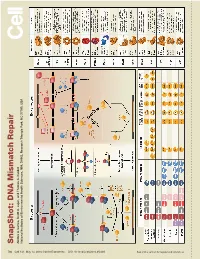
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Use of CRISPR-Modified Human Stem Cell Organoids to Study the Origin Of
RESEARCH CANCER MLH1 in normal human colonic organoids, we used CRISPR-Cas9 technology to insert a puromycin- resistance cassette into the second exon of the MLH1 gene (Fig. 1A and fig. S1A). After puromycin Use of CRISPR-modified human stem selection, we clonally expanded single organoids and subsequently genotyped these to confirm cor- cell organoids to study the origin of rect biallelic targeting (figs. S1, A and B, and S2A). Gene inactivation was verified by quantitative mutational signatures in cancer reverse transcription polymerase chain reaction (qRT-PCR), revealing a substantial reduction in MLH1 mRNA expression in MLH1 knockout Jarno Drost,1,2*† Ruben van Boxtel,2,3*† Francis Blokzijl,2,3 Tomohiro Mizutani,1,2 (MLH1KO) organoids, most likely due to the deg- Nobuo Sasaki,1,2 Valentina Sasselli,1,2 Joep de Ligt,2,3 Sam Behjati,4,5 radation (through nonsense-mediated decay) of Judith E. Grolleman,6 Tom van Wezel,7 Serena Nik-Zainal,4,8 Roland P. Kuiper,6,9 nonsense mRNAs transcribed from the mutant Edwin Cuppen,2,3‡ Hans Clevers1,2,9‡ alleles (Fig. 1C). Western blot analysis confirmed the loss of MLH1 protein expression in the MLH1KO Mutational processes underlie cancer initiation and progression. Signatures of these processes organoids (Fig. 1E). in cancer genomes may explain cancer etiology and could hold diagnostic and prognostic Next, we passaged the MLH1KO and parental value. We developed a strategy that can be used to explore the origin of cancer-associated normal human colon organoids for 2 months to mutational signatures. We used CRISPR-Cas9 technology to delete key DNA repair genes allow cells to accumulate sufficient mutations re- in human colon organoids, followed by delayed subcloning and whole-genome sequencing. -

The Properties of Msh2–Msh6 ATP Binding Mutants Suggest a Signal
ARTICLE cro Author’s Choice The properties of Msh2–Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair Received for publication, August 19, 2018, and in revised form, September 17, 2018 Published, Papers in Press, September 20, 2018, DOI 10.1074/jbc.RA118.005439 William J. Graham, V‡, Christopher D. Putnam‡§, and X Richard D. Kolodner‡¶ʈ**1 From the ‡Ludwig Institute for Cancer Research San Diego, Departments of §Medicine and ¶Cellular and Molecular Medicine, ʈMoores-UCSD Cancer Center, and **Institute of Genomic Medicine, University of California School of Medicine, San Diego, La Jolla, California 92093-0669 Edited by Patrick Sung DNA mismatch repair (MMR) corrects mispaired DNA bases synthesis (1–6). MMR also acts on some forms of chemically and small insertion/deletion loops generated by DNA replica- damaged DNA bases as well as on mispairs present in hetero- tion errors. After binding a mispair, the eukaryotic mispair rec- duplex DNA intermediates formed during recombination ognition complex Msh2–Msh6 binds ATP in both of its nucle- (1–6). MMR proteins also act in a pathway that suppresses otide-binding sites, which induces a conformational change recombination between homologous but divergent DNAs resulting in the formation of an Msh2–Msh6 sliding clamp that (1–6) and function in a DNA damage response pathway for releases from the mispair and slides freely along the DNA. How- different types of DNA-damaging agents, including some che- ever, the roles that Msh2–Msh6 sliding clamps play in MMR motherapeutic agents (2). As a result of the role that MMR plays remain poorly understood.