Germline and Tumor Whole Genome Sequencing As a Diagnostic Tool to Resolve Suspected Lynch Syndrome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Trajectory and Uniqueness of Mutational Signatures in Yeast Mutators
Trajectory and uniqueness of mutational signatures in yeast mutators Sophie Loeilleta,b, Mareike Herzogc, Fabio Pudduc, Patricia Legoixd, Sylvain Baulanded, Stephen P. Jacksonc, and Alain G. Nicolasa,b,1 aInstitut Curie, Paris Sciences et Lettres Research University, CNRS, UMR3244, 75248 Paris Cedex 05, France; bSorbonne Universités, Université Pierre et Marie Curie Paris 06, CNRS, UMR3244, 75248 Paris Cedex 05, France; cWellcome/Cancer Research UK Gurdon Institute and Department of Biochemistry, Cambridge CB2 1QN, United Kingdom; and dICGex NGS Platform, Institut Curie, 75248 Paris Cedex 05, France Edited by Richard D. Kolodner, Ludwig Institute for Cancer Research, La Jolla, CA, and approved August 24, 2020 (received for review June 2, 2020) The acquisition of mutations plays critical roles in adaptation, evolution, known (5, 15, 16). Here, we conducted a reciprocal functional ap- senescence, and tumorigenesis. Massive genome sequencing has proach to inactivate one or several genes involved in distinct ge- allowed extraction of specific features of many mutational landscapes nome maintenance processes (replication, repair, recombination, but it remains difficult to retrospectively determine the mechanistic oxidative stress response, or cell-cycle progression) in Saccharomy- origin(s), selective forces, and trajectories of transient or persistent ces cerevisiae diploids, establish the genome-wide mutational land- mutations and genome rearrangements. Here, we conducted a pro- scapes of mutation accumulation (MA) lines, explore the underlying spective reciprocal approach to inactivate 13 single or multiple evolu- mechanisms, and characterize the dynamics of mutation accumu- tionary conserved genes involved in distinct genome maintenance lation (and disappearance) along single-cell bottleneck passages. processes and characterize de novo mutations in 274 diploid Saccharo- myces cerevisiae mutation accumulation lines. -

3256.Full.Pdf
Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair Julie Akiko Heck, Juan Lucas Argueso, Zekeriyya Gemici, Richard Guy Reeves*, Ann Bernard, Charles F. Aquadro, and Eric Alani† Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853 Communicated by Thomas D. Petes, University of North Carolina, Chapel Hill, NC, December 20, 2005 (received for review June 22, 2005) In budding yeast, the MLH1-PMS1 heterodimer is the major MutL The influences of epistatic interactions on a wide variety of homolog complex that acts to repair mismatches arising during traits and processes have garnered increasing attention (14–17). DNA replication. Using a highly sensitive mutator assay, we ob- Few examples, however, have been characterized in molecular served that Saccharomyces cerevisiae strains bearing the S288c- detail. Here we show that the strain-dependent MMR pheno- strain-derived MLH1 gene and the SK1-strain-derived PMS1 gene types observed previously for site-specific mlh1 mutants were displayed elevated mutation rates that conferred a long-term due in part to an underlying defect between wild-type MMR fitness cost. Dissection of this negative epistatic interaction using genes from S288c and SK1. We identified the specific amino acid S288c-SK1 chimeras revealed that a single amino acid polymor- polymorphisms in MLH1 and PMS1, whose combined effect in phism in each gene accounts for this mismatch repair defect. Were hybrid strains leads to an elevation in mutation rate and a these strains to cross in natural populations, segregation of alleles generalized reduction in long-term fitness. -

DF6906-PMS1 Antibody
Affinity Biosciences website:www.affbiotech.com order:[email protected] PMS1 Antibody Cat.#: DF6906 Concn.: 1mg/ml Mol.Wt.: 106kDa Size: 50ul,100ul,200ul Source: Rabbit Clonality: Polyclonal Application: WB 1:500-1:2000, IHC 1:50-1:200, ELISA(peptide) 1:20000-1:40000 *The optimal dilutions should be determined by the end user. Reactivity: Human,Rat Purification: The antiserum was purified by peptide affinity chromatography using SulfoLink™ Coupling Resin (Thermo Fisher Scientific). Specificity: PMS1 Antibody detects endogenous levels of total PMS1. Immunogen: A synthesized peptide derived from human PMS1, corresponding to a region within the internal amino acids. Uniprot: P54277 Description: PMS1 belongs to the DNA mismatch repair mutL/hexB family. It is thought to be involved in the repair of DNA mismatches, and it can form heterodimers with MLH1, a known DNA mismatch repair protein. Mutations in PMS1 cause hereditary nonpolyposis colorectal cancer type 3 (HNPCC3) either alone or in combination with mutations in other proteins involved in the HNPCC phenotype, which is also known as Lynch syndrome (1). Storage Condition and Rabbit IgG in phosphate buffered saline , pH 7.4, 150mM Buffer: NaCl, 0.02% sodium azide and 50% glycerol.Store at -20 °C.Stable for 12 months from date of receipt. Western blot analysis of Hela whole cell lysates, using PMS1 Antibody. The lane on the left was treated with the antigen- specific peptide. 1 / 2 Affinity Biosciences website:www.affbiotech.com order:[email protected] DF6906 at 1/100 staining Human esophageal cancer by IHC-P. The sample was formaldehyde fixed and a heat mediated antigen retrieval step in citrate buffer was performed. -

Negative Epistasis Between Natural Variants of the Saccharomyces Cerevisiae MLH1 and PMS1 Genes Results in a Defect in Mismatch Repair
Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair Julie Akiko Heck, Juan Lucas Argueso, Zekeriyya Gemici, Richard Guy Reeves*, Ann Bernard, Charles F. Aquadro, and Eric Alani† Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853 Communicated by Thomas D. Petes, University of North Carolina, Chapel Hill, NC, December 20, 2005 (received for review June 22, 2005) In budding yeast, the MLH1-PMS1 heterodimer is the major MutL The influences of epistatic interactions on a wide variety of homolog complex that acts to repair mismatches arising during traits and processes have garnered increasing attention (14–17). DNA replication. Using a highly sensitive mutator assay, we ob- Few examples, however, have been characterized in molecular served that Saccharomyces cerevisiae strains bearing the S288c- detail. Here we show that the strain-dependent MMR pheno- strain-derived MLH1 gene and the SK1-strain-derived PMS1 gene types observed previously for site-specific mlh1 mutants were displayed elevated mutation rates that conferred a long-term due in part to an underlying defect between wild-type MMR fitness cost. Dissection of this negative epistatic interaction using genes from S288c and SK1. We identified the specific amino acid S288c-SK1 chimeras revealed that a single amino acid polymor- polymorphisms in MLH1 and PMS1, whose combined effect in phism in each gene accounts for this mismatch repair defect. Were hybrid strains leads to an elevation in mutation rate and a these strains to cross in natural populations, segregation of alleles generalized reduction in long-term fitness. -

Conformational Trapping of Mismatch Recognition Complex MSH2/MSH3
Conformational trapping of Mismatch Recognition PNAS PLUS Complex MSH2/MSH3 on repair-resistant DNA loops Walter H. Langa,1,2, Julie E. Coatsb,1, Jerzy Majkaa, Greg L. Huraa, Yuyen Linb, Ivan Rasnikb,3, and Cynthia T. McMurraya,c,d,3 aLawrence Berkeley National Laboratory, Life Sciences Division, 1 Cyclotron Road, Berkeley, CA 94720; cDepartment of Molecular Pharmacology and Experimental Therapeutics; dDepartment of Biochemistry and Molecular Biology, Mayo Foundation, 200 First Street, Rochester, MN 55905; and bDepartment of Physics, Emory University, 400 Dowman Drive, MSC N214, Atlanta, GA 30322 Edited* by Peter H. von Hippel, Institute of Molecular Biology, Eugene, OR, and approved August 3, 2011 (received for review April 6, 2011) Insertion and deletion of small heteroduplex loops are common which fails to effectively couple DNA binding with downstream mutations in DNA, but why some loops are prone to mutation and repair signaling. We envision that conformational regulation of others are efficiently repaired is unknown. Here we report that the small loop repair occurs at the level of the junction dynamics. mismatch recognition complex, MSH2/MSH3, discriminates between a repair-competent and a repair-resistant loop by sensing the con- Results formational dynamics of their junctions. MSH2/MSH3 binds, bends, Conformational Integrity of MSH2/MSH3 and the DNA “Junction” Tem- and dissociates from repair-competent loops to signal downstream plates. We characterized the DNA-binding affinity and nucleotide repair. Repair-resistant Cytosine-Adenine-Guanine (CAG) loops adopt binding properties of MSH2/MSH3 bound to looped templates, a unique DNA junction that traps nucleotide-bound MSH2/MSH3, either a ðCAÞ4 loop or CAG hairpin loops of either 7 or 13 and inhibits its dissociation from the DNA. -

Dihydrofolate Reductase Amplification and Sensitization to Methotrexate of Methotrexate-Resistant Colon Cancer Cells
Published OnlineFirst February 3, 2009; DOI: 10.1158/1535-7163.MCT-08-0759 424 Dihydrofolate reductase amplification and sensitization to methotrexate of methotrexate-resistant colon cancer cells Cristina Morales,1 Maria J. Garcı´a,4 Maria Ribas,1 withdrawal and reexposure to MTX. Passive loss of the Rosa Miro´,2 Mar Mun˜oz,3 Carlos Caldas,4 DHFR amplicon by withdrawal of the drug results in MTX- and Miguel A. Peinado1,3 sensitive cells exhibiting a substantial reduction of their capacity or even an incapacity to generate resistance when 1Institut d’Investigacio´Biome`dica de Bellvitge, L’Hospitalet; submitted to a second cycle of MTX treatment. On a 2 Institut de Biotecnologia i Biomedicina, Departament de Biologia second round of drug administration, the resistant cells CelÁlular, Fisiologia i Immunologia, Universitat Autonoma de Barcelona, Bellaterra; 3Institut de Medicina Predictiva i generate a different amplicon structure, suggesting that the Personalitzada del Ca`ncer, Badalona, Barcelona, Spain and formation of the amplicon as in the first cycle of treatment 4Breast Cancer Functional Genomics Laboratory, Cancer is not feasible. These results indicate that DHFR gene Research UK Cambridge Research Institute and Department of amplification is a ‘‘wear and tear’’ process in HT29 cells Oncology, University of Cambridge, Li Ka Shing Centre, and that MTX-resistant cells may become responsive to a Cambridge, United Kingdom second round of treatment if left untreated during a sufficient period of time. [Mol Cancer Ther 2009; Abstract 8(2):424–32] Gene amplification is one of the most frequent manifes- tations of genomic instability in human tumors and plays an important role in tumor progression and acquisition of Introduction drug resistance. -

Recruitment of Mismatch Repair Proteins to the Site of DNA Damage in Human Cells
3146 Research Article Recruitment of mismatch repair proteins to the site of DNA damage in human cells Zehui Hong, Jie Jiang, Kazunari Hashiguchi, Mikiko Hoshi, Li Lan and Akira Yasui* Department of Molecular Genetics, Institute of Development, Aging and Cancer, Tohoku University, Seiryomachi 4-1, Aobaku, Sendai 980-8575, Japan *Author for correspondence (e-mail: [email protected]) Accepted 8 July 2008 Journal of Cell Science 121, 3146-3154 Published by The Company of Biologists 2008 doi:10.1242/jcs.026393 Summary Mismatch repair (MMR) proteins contribute to genome stability the PCNA-binding domain of MSH6. MSH2 is recruited to the by excising DNA mismatches introduced by DNA polymerase. DNA damage site through interactions with either MSH3 or Although MMR proteins are also known to influence cellular MSH6, and is required for recruitment of MLH1 to the damage responses to DNA damage, how MMR proteins respond to DNA site. We found, furthermore, that MutSβ is also recruited to damage within the cell remains unknown. Here, we show that UV-irradiated sites in nucleotide-excision-repair- and PCNA- MMR proteins are recruited immediately to the sites of various dependent manners. Thus, MMR and its proteins function not types of DNA damage in human cells. MMR proteins are only in replication but also in DNA repair. recruited to single-strand breaks in a poly(ADP-ribose)- dependent manner as well as to double-strand breaks. Using Supplementary material available online at mutant cells, RNA interference and expression of fluorescence- http://jcs.biologists.org/cgi/content/full/121/19/3146/DC1 -

PMS2 Antibody / PMS1 Homolog 2 (RQ4366)
PMS2 Antibody / PMS1 homolog 2 (RQ4366) Catalog No. Formulation Size RQ4366 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Antigen affinity purified Buffer Lyophilized from 1X PBS with 2% Trehalose and 0.025% sodium azide UniProt P54278 Localization Nucleus Applications Western blot : 0.5-1ug/ml Direct ELISA : 0.1-0.5ug/ml Limitations This PMS2 antibody is available for research use only. Western blot testing of human HeLa cell lysate with BCMA antibody at 0.5ug/ml. Expected molecular weight: 96-110 kDa. Description Mismatch repair endonuclease PMS2 is an enzyme that in humans is encoded by the PMS2 gene. The protein encoded by this gene is a key component of the mismatch repair system that functions to correct DNA mismatches and small insertions and deletions that can occur during DNA replication and homologous recombination. This protein forms heterodimers with the gene product of the mutL homolog 1 (MLH1) gene to form the MutL-alpha heterodimer. The MutL-alpha heterodimer possesses an endonucleolytic activity that is activated following recognition of mismatches and insertion/deletion loops by the MutS-alpha and MutS-beta heterodimers, and is necessary for removal of the mismatched DNA. There is a DQHA(X)2E(X)4E motif found at the C-terminus of the protein encoded by this gene that forms part of the active site of the nuclease. Mutations in this gene have been associated with hereditary nonpolyposis colorectal cancer (HNPCC; also known as Lynch syndrome) and Turcot syndrome. -
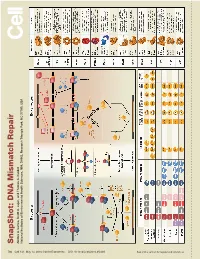
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

Machine Learning-Based Approach Highlights the Use of a Genomic Variant Profile for Precision Medicine in Ovarian Failure
Journal of Personalized Medicine Article Machine Learning-Based Approach Highlights the Use of a Genomic Variant Profile for Precision Medicine in Ovarian Failure Ismael Henarejos-Castillo 1,2 , Alejandro Aleman 1, Begoña Martinez-Montoro 3, Francisco Javier Gracia-Aznárez 4, Patricia Sebastian-Leon 1,3, Monica Romeu 5, Jose Remohi 2,6, Ana Patiño-Garcia 4,7 , Pedro Royo 3, Gorka Alkorta-Aranburu 4 and Patricia Diaz-Gimeno 1,3,* 1 IVI Foundation-Instituto de Investigación Sanitaria La Fe, Av. Fernando Abril Martorell 106, Torre A, Planta 1ª, 46026 Valencia, Spain; [email protected] (I.H.-C.); [email protected] (A.A.); [email protected] (P.S.-L.) 2 Department of Paediatrics, Obstetrics and Gynaecology, University of Valencia, Av. Blasco Ibáñez 15, 46010 Valencia, Spain; [email protected] 3 IVI-RMA Pamplona, Reproductive Medicine, C/Sangüesa, Número 15-Planta Baja, 31003 Pamplona, Spain; [email protected] (B.M.-M.); [email protected] (P.R.) 4 CIMA Lab Diagnostics, University of Navarra, IdiSNA, Avda Pio XII, 55, 31008 Pamplona, Spain; [email protected] (F.J.G.-A.); [email protected] (A.P.-G.); [email protected] (G.A.-A.) 5 Hospital Universitario y Politécnico La Fe, Av. Fernando Abril Martorell 106, 46026 Valencia, Spain; [email protected] 6 IVI-RMA Valencia, Reproductive Medicine, Plaça de la Policia Local, 3, 46015 Valencia, Spain 7 Laboratorio de Pediatría-Unidad de Genética Clínica, Clínica Universidad de Navarra, Avda Pio XII, 55, Citation: Henarejos-Castillo, I.; 31008 Pamplona, Spain Aleman, A.; Martinez-Montoro, B.; * Correspondence: [email protected] Gracia-Aznárez, F.J.; Sebastian-Leon, P.; Romeu, M.; Remohi, J.; Patiño- Abstract: Ovarian failure (OF) is a common cause of infertility usually diagnosed as idiopathic, Garcia, A.; Royo, P.; Alkorta-Aranburu, with genetic causes accounting for 10–25% of cases. -

The Properties of Msh2–Msh6 ATP Binding Mutants Suggest a Signal
ARTICLE cro Author’s Choice The properties of Msh2–Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair Received for publication, August 19, 2018, and in revised form, September 17, 2018 Published, Papers in Press, September 20, 2018, DOI 10.1074/jbc.RA118.005439 William J. Graham, V‡, Christopher D. Putnam‡§, and X Richard D. Kolodner‡¶ʈ**1 From the ‡Ludwig Institute for Cancer Research San Diego, Departments of §Medicine and ¶Cellular and Molecular Medicine, ʈMoores-UCSD Cancer Center, and **Institute of Genomic Medicine, University of California School of Medicine, San Diego, La Jolla, California 92093-0669 Edited by Patrick Sung DNA mismatch repair (MMR) corrects mispaired DNA bases synthesis (1–6). MMR also acts on some forms of chemically and small insertion/deletion loops generated by DNA replica- damaged DNA bases as well as on mispairs present in hetero- tion errors. After binding a mispair, the eukaryotic mispair rec- duplex DNA intermediates formed during recombination ognition complex Msh2–Msh6 binds ATP in both of its nucle- (1–6). MMR proteins also act in a pathway that suppresses otide-binding sites, which induces a conformational change recombination between homologous but divergent DNAs resulting in the formation of an Msh2–Msh6 sliding clamp that (1–6) and function in a DNA damage response pathway for releases from the mispair and slides freely along the DNA. How- different types of DNA-damaging agents, including some che- ever, the roles that Msh2–Msh6 sliding clamps play in MMR motherapeutic agents (2). As a result of the role that MMR plays remain poorly understood.