Homework Project – Protein Structure Prediction Contact Me If You Have Any Questions/Problems with the Assignment: [email protected]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Trajectory and Uniqueness of Mutational Signatures in Yeast Mutators
Trajectory and uniqueness of mutational signatures in yeast mutators Sophie Loeilleta,b, Mareike Herzogc, Fabio Pudduc, Patricia Legoixd, Sylvain Baulanded, Stephen P. Jacksonc, and Alain G. Nicolasa,b,1 aInstitut Curie, Paris Sciences et Lettres Research University, CNRS, UMR3244, 75248 Paris Cedex 05, France; bSorbonne Universités, Université Pierre et Marie Curie Paris 06, CNRS, UMR3244, 75248 Paris Cedex 05, France; cWellcome/Cancer Research UK Gurdon Institute and Department of Biochemistry, Cambridge CB2 1QN, United Kingdom; and dICGex NGS Platform, Institut Curie, 75248 Paris Cedex 05, France Edited by Richard D. Kolodner, Ludwig Institute for Cancer Research, La Jolla, CA, and approved August 24, 2020 (received for review June 2, 2020) The acquisition of mutations plays critical roles in adaptation, evolution, known (5, 15, 16). Here, we conducted a reciprocal functional ap- senescence, and tumorigenesis. Massive genome sequencing has proach to inactivate one or several genes involved in distinct ge- allowed extraction of specific features of many mutational landscapes nome maintenance processes (replication, repair, recombination, but it remains difficult to retrospectively determine the mechanistic oxidative stress response, or cell-cycle progression) in Saccharomy- origin(s), selective forces, and trajectories of transient or persistent ces cerevisiae diploids, establish the genome-wide mutational land- mutations and genome rearrangements. Here, we conducted a pro- scapes of mutation accumulation (MA) lines, explore the underlying spective reciprocal approach to inactivate 13 single or multiple evolu- mechanisms, and characterize the dynamics of mutation accumu- tionary conserved genes involved in distinct genome maintenance lation (and disappearance) along single-cell bottleneck passages. processes and characterize de novo mutations in 274 diploid Saccharo- myces cerevisiae mutation accumulation lines. -

3256.Full.Pdf
Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair Julie Akiko Heck, Juan Lucas Argueso, Zekeriyya Gemici, Richard Guy Reeves*, Ann Bernard, Charles F. Aquadro, and Eric Alani† Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853 Communicated by Thomas D. Petes, University of North Carolina, Chapel Hill, NC, December 20, 2005 (received for review June 22, 2005) In budding yeast, the MLH1-PMS1 heterodimer is the major MutL The influences of epistatic interactions on a wide variety of homolog complex that acts to repair mismatches arising during traits and processes have garnered increasing attention (14–17). DNA replication. Using a highly sensitive mutator assay, we ob- Few examples, however, have been characterized in molecular served that Saccharomyces cerevisiae strains bearing the S288c- detail. Here we show that the strain-dependent MMR pheno- strain-derived MLH1 gene and the SK1-strain-derived PMS1 gene types observed previously for site-specific mlh1 mutants were displayed elevated mutation rates that conferred a long-term due in part to an underlying defect between wild-type MMR fitness cost. Dissection of this negative epistatic interaction using genes from S288c and SK1. We identified the specific amino acid S288c-SK1 chimeras revealed that a single amino acid polymor- polymorphisms in MLH1 and PMS1, whose combined effect in phism in each gene accounts for this mismatch repair defect. Were hybrid strains leads to an elevation in mutation rate and a these strains to cross in natural populations, segregation of alleles generalized reduction in long-term fitness. -

DF6906-PMS1 Antibody
Affinity Biosciences website:www.affbiotech.com order:[email protected] PMS1 Antibody Cat.#: DF6906 Concn.: 1mg/ml Mol.Wt.: 106kDa Size: 50ul,100ul,200ul Source: Rabbit Clonality: Polyclonal Application: WB 1:500-1:2000, IHC 1:50-1:200, ELISA(peptide) 1:20000-1:40000 *The optimal dilutions should be determined by the end user. Reactivity: Human,Rat Purification: The antiserum was purified by peptide affinity chromatography using SulfoLink™ Coupling Resin (Thermo Fisher Scientific). Specificity: PMS1 Antibody detects endogenous levels of total PMS1. Immunogen: A synthesized peptide derived from human PMS1, corresponding to a region within the internal amino acids. Uniprot: P54277 Description: PMS1 belongs to the DNA mismatch repair mutL/hexB family. It is thought to be involved in the repair of DNA mismatches, and it can form heterodimers with MLH1, a known DNA mismatch repair protein. Mutations in PMS1 cause hereditary nonpolyposis colorectal cancer type 3 (HNPCC3) either alone or in combination with mutations in other proteins involved in the HNPCC phenotype, which is also known as Lynch syndrome (1). Storage Condition and Rabbit IgG in phosphate buffered saline , pH 7.4, 150mM Buffer: NaCl, 0.02% sodium azide and 50% glycerol.Store at -20 °C.Stable for 12 months from date of receipt. Western blot analysis of Hela whole cell lysates, using PMS1 Antibody. The lane on the left was treated with the antigen- specific peptide. 1 / 2 Affinity Biosciences website:www.affbiotech.com order:[email protected] DF6906 at 1/100 staining Human esophageal cancer by IHC-P. The sample was formaldehyde fixed and a heat mediated antigen retrieval step in citrate buffer was performed. -

Negative Epistasis Between Natural Variants of the Saccharomyces Cerevisiae MLH1 and PMS1 Genes Results in a Defect in Mismatch Repair
Negative epistasis between natural variants of the Saccharomyces cerevisiae MLH1 and PMS1 genes results in a defect in mismatch repair Julie Akiko Heck, Juan Lucas Argueso, Zekeriyya Gemici, Richard Guy Reeves*, Ann Bernard, Charles F. Aquadro, and Eric Alani† Department of Molecular Biology and Genetics, Cornell University, Ithaca, NY 14853 Communicated by Thomas D. Petes, University of North Carolina, Chapel Hill, NC, December 20, 2005 (received for review June 22, 2005) In budding yeast, the MLH1-PMS1 heterodimer is the major MutL The influences of epistatic interactions on a wide variety of homolog complex that acts to repair mismatches arising during traits and processes have garnered increasing attention (14–17). DNA replication. Using a highly sensitive mutator assay, we ob- Few examples, however, have been characterized in molecular served that Saccharomyces cerevisiae strains bearing the S288c- detail. Here we show that the strain-dependent MMR pheno- strain-derived MLH1 gene and the SK1-strain-derived PMS1 gene types observed previously for site-specific mlh1 mutants were displayed elevated mutation rates that conferred a long-term due in part to an underlying defect between wild-type MMR fitness cost. Dissection of this negative epistatic interaction using genes from S288c and SK1. We identified the specific amino acid S288c-SK1 chimeras revealed that a single amino acid polymor- polymorphisms in MLH1 and PMS1, whose combined effect in phism in each gene accounts for this mismatch repair defect. Were hybrid strains leads to an elevation in mutation rate and a these strains to cross in natural populations, segregation of alleles generalized reduction in long-term fitness. -

PMS2 Antibody / PMS1 Homolog 2 (RQ4366)
PMS2 Antibody / PMS1 homolog 2 (RQ4366) Catalog No. Formulation Size RQ4366 0.5mg/ml if reconstituted with 0.2ml sterile DI water 100 ug Bulk quote request Availability 1-3 business days Species Reactivity Human Format Antigen affinity purified Clonality Polyclonal (rabbit origin) Isotype Rabbit IgG Purity Antigen affinity purified Buffer Lyophilized from 1X PBS with 2% Trehalose and 0.025% sodium azide UniProt P54278 Localization Nucleus Applications Western blot : 0.5-1ug/ml Direct ELISA : 0.1-0.5ug/ml Limitations This PMS2 antibody is available for research use only. Western blot testing of human HeLa cell lysate with BCMA antibody at 0.5ug/ml. Expected molecular weight: 96-110 kDa. Description Mismatch repair endonuclease PMS2 is an enzyme that in humans is encoded by the PMS2 gene. The protein encoded by this gene is a key component of the mismatch repair system that functions to correct DNA mismatches and small insertions and deletions that can occur during DNA replication and homologous recombination. This protein forms heterodimers with the gene product of the mutL homolog 1 (MLH1) gene to form the MutL-alpha heterodimer. The MutL-alpha heterodimer possesses an endonucleolytic activity that is activated following recognition of mismatches and insertion/deletion loops by the MutS-alpha and MutS-beta heterodimers, and is necessary for removal of the mismatched DNA. There is a DQHA(X)2E(X)4E motif found at the C-terminus of the protein encoded by this gene that forms part of the active site of the nuclease. Mutations in this gene have been associated with hereditary nonpolyposis colorectal cancer (HNPCC; also known as Lynch syndrome) and Turcot syndrome. -
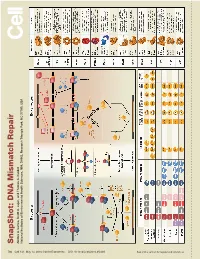
S Na P S H O T: D N a Mism a Tc H R E P a Ir
SnapShot: Repair DNA Mismatch Scott A. Lujan, and Thomas Kunkel A. Larrea, Andres Park, NC 27709, USA Triangle Health Sciences, NIH, DHHS, Research National Institutes of Environmental 730 Cell 141, May 14, 2010 ©2010 Elsevier Inc. DOI 10.1016/j.cell.2010.05.002 See online version for legend and references. SnapShot: DNA Mismatch Repair Andres A. Larrea, Scott A. Lujan, and Thomas A. Kunkel National Institutes of Environmental Health Sciences, NIH, DHHS, Research Triangle Park, NC 27709, USA Mismatch Repair in Bacteria and Eukaryotes Mismatch repair in the bacterium Escherichia coli is initiated when a homodimer of MutS binds as an asymmetric clamp to DNA containing a variety of base-base and insertion-deletion mismatches. The MutL homodimer then couples MutS recognition to the signal that distinguishes between the template and nascent DNA strands. In E. coli, the lack of adenine methylation, catalyzed by the DNA adenine methyltransferase (Dam) in newly synthesized GATC sequences, allows E. coli MutH to cleave the nascent strand. The resulting nick is used for mismatch removal involving the UvrD helicase, single-strand DNA-binding protein (SSB), and excision by single-stranded DNA exonucleases from either direction, depending upon the polarity of the nick relative to the mismatch. DNA polymerase III correctly resynthesizes DNA and ligase completes repair. In bacteria lacking Dam/MutH, as in eukaryotes, the signal for strand discrimination is uncertain but may be the DNA ends associated with replication forks. In these bacteria, MutL harbors a nick-dependent endonuclease that creates a nick that can be used for mismatch excision. Eukaryotic mismatch repair is similar, although it involves several dif- ferent MutS and MutL homologs: MutSα (MSH2/MSH6) recognizes single base-base mismatches and 1–2 base insertion/deletions; MutSβ (MSH2/MSH3) recognizes insertion/ deletion mismatches containing two or more extra bases. -

The Properties of Msh2–Msh6 ATP Binding Mutants Suggest a Signal
ARTICLE cro Author’s Choice The properties of Msh2–Msh6 ATP binding mutants suggest a signal amplification mechanism in DNA mismatch repair Received for publication, August 19, 2018, and in revised form, September 17, 2018 Published, Papers in Press, September 20, 2018, DOI 10.1074/jbc.RA118.005439 William J. Graham, V‡, Christopher D. Putnam‡§, and X Richard D. Kolodner‡¶ʈ**1 From the ‡Ludwig Institute for Cancer Research San Diego, Departments of §Medicine and ¶Cellular and Molecular Medicine, ʈMoores-UCSD Cancer Center, and **Institute of Genomic Medicine, University of California School of Medicine, San Diego, La Jolla, California 92093-0669 Edited by Patrick Sung DNA mismatch repair (MMR) corrects mispaired DNA bases synthesis (1–6). MMR also acts on some forms of chemically and small insertion/deletion loops generated by DNA replica- damaged DNA bases as well as on mispairs present in hetero- tion errors. After binding a mispair, the eukaryotic mispair rec- duplex DNA intermediates formed during recombination ognition complex Msh2–Msh6 binds ATP in both of its nucle- (1–6). MMR proteins also act in a pathway that suppresses otide-binding sites, which induces a conformational change recombination between homologous but divergent DNAs resulting in the formation of an Msh2–Msh6 sliding clamp that (1–6) and function in a DNA damage response pathway for releases from the mispair and slides freely along the DNA. How- different types of DNA-damaging agents, including some che- ever, the roles that Msh2–Msh6 sliding clamps play in MMR motherapeutic agents (2). As a result of the role that MMR plays remain poorly understood. -

Abstract the Human Protein PMS1 Is a Protein That Functions in DNA Mismatch Repair
ii iv Abstract The human protein PMS1 is a protein that functions in DNA mismatch repair. PMS1 is part of the High Motility Group Protein family (HMG proteins). Using homology modeling in YASARA, a 3-dimensional structure of the PMS1 protein was produced and the structure was verified as realistic using molecular dynamics. Using Evolutionary Analysis in an online program called ConSurf identified the high and low conservative regions of the PMS1 protein. Sequences with high conservation scores indicate important structural and functional aspects of the proteins. Using the GNOMAD database, human variants of the protein were found with a focus placed on those that caused missense, loss of function and frameshift mutations which can be found in the 3-dimensional structure. Where these proteins are found will be at the sites of phenotypical consequences from mutation. Using the Human Protein Atlas, PMS1 was found in all cells. However, it is most common in cells with a high reproductivity rate, like cells in the digestive tract. Malfunctioning of PMS1 leads to genome instability and more frequent mutations, which can cause genetic defects or cancers. The Catalogue of Somatic Mutation in Cancer (COSMIC) was used to identify cancer related mutations of PMS1, such as colorectal cancer. It is hoped that this sequence to structure to function to phenotype approach will contribute to the future of genomic medicine. v Acknowledgments I would like to thank Dr. Hawkins and the entire honors committee for the riveting opportunity to partake in Walsh University’s Honors Program. This faculty has pushed me past my comfort level for the past four years to become the successful student and person I am today. -

Germline and Tumor Whole Genome Sequencing As a Diagnostic Tool to Resolve Suspected Lynch Syndrome
medRxiv preprint doi: https://doi.org/10.1101/2020.03.12.20034991; this version posted March 17, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. All rights reserved. No reuse allowed without permission. Germline and Tumor Whole Genome Sequencing as a Diagnostic Tool to Resolve Suspected Lynch Syndrome Bernard J. Pope1,2,3, Mark Clendenning1,2, Christophe Rosty1,2,4,5, Khalid Mahmood1,2,3, Peter Georgeson1,2, Jihoon E. Joo1,2, Romy Walker1,2, Ryan A. Hutchinson1,2, Harindra Jayasekara1,2,6, Sharelle Joseland1,2, Julia Como1,2, Susan Preston1,2, Amanda B. Spurdle7, Finlay A. Macrae8,9,10, Aung K. Win11, John L. Hopper11, Mark A. Jenkins11, Ingrid M. Winship9,10, Daniel D. Buchanan1,2,10* 1 Colorectal Oncogenomics Group, Department of Clinical Pathology, The University of Melbourne, Parkville, Victoria, Australia 2 University of Melbourne Centre for Cancer Research, Victorian Comprehensive Cancer Centre, Parkville, Victoria, Australia 3 Melbourne Bioinformatics, The University of Melbourne, Parkville, Victoria, Australia 4 Envoi Pathology, Brisbane, Queensland, Australia 5 University of Queensland, School of Medicine, Herston, Queensland, Australia 6 Cancer Epidemiology Division, Cancer Council Victoria, Melbourne, Victoria, Australia 7 Molecular Cancer Epidemiology Laboratory, QIMR Berghofer Medical Research Institute, Herston, Brisbane, Queensland, Australia 8 Colorectal Medicine and Genetics, The Royal Melbourne -

Analysis of DNA Mismatch Repair Gene Expression and Mutations in Thyroid Tumours
ANTICANCER RESEARCH 26: 2107-2112 (2006) Analysis of DNA Mismatch Repair Gene Expression and Mutations in Thyroid Tumours ILKA RUSCHENBURG1, BEATE VOLLHEIM2, JERZY STACHURA3, CARLOS CORDON-CARDO4 and MONIKA KORABIOWSKA2 1Cytological Laboratory, Einbeck; 2Department of Pathology, Reinhard Nieter Hospital, Academic Hospital of the University of Göttingen, Wilhelmshaven, Germany; 3Department of Pathology, Jagiellonian University Krakow, Poland; 4Division of Molecular Pathology, Memorial Sloan Kettering Cancer Center, New York, U.S.A. Abstract. Alterations of DNA mismatch repair genes, Microsatellite instability and some LOH have been found primarily demonstrated in hereditary nonpolyposis colorectal in thyroid carcinomas (13, 14). Accepting microsatellite carcinomas, were reported to be of relevance for the instability as the result of malfunctioning DNA mismatch progression of several sporadic tumours. In this study, the repair, a working hypothesis that some alterations of DNA expression and mutations of MLH1, MSH2, PMS1 and PMS2 mismatch repair protein expression and eventual mutations in a panel of thyroid tumours, including nodular hyperplasia, in DNA mismatch repair genes are found in thyroid follicular adenomas and carcinomas, were investigated. The carcinomas can be formulated. Deletion of the short arm expressions of MLH1, MSH2 and PMS1 were generally higher of chromosome 3, including the locus of MLH1, reported in malignant tumours than in benign lesions (p<0.01). This for thyroid carcinomas, was an additional argument in observation can find potential diagnostic application in the favour of this hypothesis (15). The overall target of this differentiation of follicular adenomas from follicullar study was to check our working hypothesis and additionally carcinomas of the thyroid. No point mutations in the DNA to judge the diagnostic relevance of DNA mismatch repair mismatch repair genes MSH2 (exon 12, 13) and MLH1 (exon gene expression. -

The Mismatch Repair System Reduces Meiotic Homeologous Recombination and Stimulates Recombination-Dependent Chromosome Loss
MOLECULAR AND CELLULAR BIOLOGY, Nov. 1996, p. 6110–6120 Vol. 16, No. 11 0270-7306/96/$04.0010 Copyright q 1996, American Society for Microbiology The Mismatch Repair System Reduces Meiotic Homeologous Recombination and Stimulates Recombination-Dependent Chromosome Loss SCOTT R. CHAMBERS, NEIL HUNTER, EDWARD J. LOUIS, AND RHONA H. BORTS* Yeast Genetics, Institute of Molecular Medicine, John Radcliffe Hospital, Oxford OX3 9DU, United Kingdom Received 6 May 1996/Returned for modification 9 July 1996/Accepted 13 August 1996 Efficient genetic recombination requires near-perfect homology between participating molecules. Sequence divergence reduces the frequency of recombination, a process that is dependent on the activity of the mismatch repair system. The effects of chromosomal divergence in diploids of Saccharomyces cerevisiae in which one copy of chromosome III is derived from a closely related species, Saccharomyces paradoxus, have been examined. Meiotic recombination between the diverged chromosomes is decreased by 25-fold. Spore viability is reduced with an observable increase in the number of tetrads with only two or three viable spores. Asci with only two viable spores are disomic for chromosome III, consistent with meiosis I nondisjunction of the homeologs. Asci with three viable spores are highly enriched for recombinants relative to tetrads with four viable spores. In 96% of the class with three viable spores, only one spore possesses a recombinant chromosome III, suggesting that the recombination process itself contributes to meiotic death. This phenomenon is dependent on the activities of the mismatch repair genes PMS1 and MSH2. A model of mismatch-stimulated chromosome loss is proposed to account for this observation.